ABSTRACT
Background: Aripiprazole lauroxil (AL) 1064 mg every 2 months following initiation using the AL NanoCrystal Dispersion formulation (ALNCD) plus 30-mg oral aripiprazole was efficacious and well tolerated in a 25-week, randomized, double-blind phase 3 trial in adults with acute schizophrenia. This post hoc analysis further characterized the safety of AL 1064 mg administered every 2 months and that of active control paliperidone palmitate (PP) 156 mg monthly based on occurrence, timing, and severity of adverse events (AEs) associated with antipsychotic medications.
Methods: This study was conducted between November 2017 and March 2019. AL or PP was initiated during an inpatient stay of ≥ 2 weeks with transition to outpatient treatment thereafter. Rates of AEs of clinical interest, including injection site reactions (ISRs), motor AEs, sedation, hypotension, prolactin level increase, weight gain, and suicidal ideation/behavior, were summarized through weeks 4, 9, and 25 for each treatment.
Results: Of 200 patients who received ≥ 1 dose of study treatment, 99 (49.5%) completed the study (AL, 57%; PP, 43%). Mean (SD) baseline Positive and Negative Syndrome Scale total scores were 94.1 (9.04) and 94.6 (8.41) in the AL and PP treatment groups, respectively. AEs were reported by 69/99 (70%) patients administered AL and 72/101 (71%) administered PP; most AEs were mild or moderate in severity. ISRs (AL, 18.2%; PP, 26.7%) occurred primarily on days 1 and 8. All akathisia/restlessness AEs (AL, 10.1%; PP, 11.9%) occurred during the first 4 weeks; <10% of patients (either treatment) experienced hypotension, sedation, or suicidal ideation/behavior events. Weight gain of ≥ 7% from baseline occurred in 9.3% of AL- and 23.8% of PP-treated patients. Median prolactin concentrations changed by −4.60 and −3.55 ng/mL among AL-treated males and females, respectively, and did not exceed 2 times normal levels in any AL-treated patients. In PP-treated patients, changes were 21.20 and 80.40 ng/mL and concentrations exceeded 2 times normal in 38% and 88% of males and females, respectively.
Conclusions: No new early- or late-emerging safety concerns were observed through 25 weeks of treatment with AL 1064 mg every 2 months following initiation using ALNCD plus 30-mg oral aripiprazole. Results were consistent with known safety profiles of AL and PP and support the safety of AL 1064 mg every 2 months initiated using ALNCD plus 30-mg oral aripiprazole.
Trial Registration: ClinicalTrials.gov identifier: NCT03345979
J Clin Psychiatry 2024;85(1):23m15095
Author affiliations are listed at the end of this article.
For patients with schizophrenia, use of antipsychotics and nonpharmacologic treatment approaches is crucial for reducing symptoms, maintaining recovery, and maximizing functioning.1,2 Antipsychotic options include oral medications administered daily and long-acting injectable (LAI) antipsychotics administered at intervals ranging from every 2 weeks to every 6 months,3,4 enabling individualization of treatment in consideration of patients’ needs, values, and preferences.1 Advantages of LAI antipsychotics include ensuring continuous medication exposure; those with longer dosing intervals require fewer injections in a given timeframe.5 However, the slow dissolution of LAI antipsychotics often necessitates additional steps to rapidly achieve therapeutic drug concentrations at initiation.6
The atypical LAI antipsychotic aripiprazole lauroxil (AL), indicated for schizophrenia in adults, is available in dosing intervals of up to 2 months’ duration.6,7 AL can be initiated in 1 day with an injection of a NanoCrystal Dispersion formulation of AL (ALNCD), the first such formulation developed specifically for LAI initiation and available as a 675-mg dose only, plus one 30-mg dose of oral aripiprazole, obviating the need for additional oral supplementation beyond day 1. The ALNCD injection and single oral aripiprazole dose together are referred to as a “1-day initiation regimen” because they can be administered on the same day as the initial maintenance AL injection. However, the product label allows flexibility; the first maintenance AL injection can be given anytime within the subsequent 10 days after ALNCD.6,8
The ALPINE (Aripiprazole Lauroxil and Paliperidone palmitate: INitiation Effectiveness) study was a randomized, active-controlled, double-blind, 25-week phase 3 trial that evaluated the efficacy and safety of every-2-month AL 1064-mg dosing initiated using ALNCD plus 30-mg oral aripiprazole for treatment of patients with an acute exacerbation of schizophrenia. Paliperidone palmitate (PP) was included as an active control.9 Patients initiated LAI antipsychotic treatment as inpatients and transitioned to outpatient care as early as day 15 if clinically stable. Each treatment resulted in significant within-group improvement in Positive and Negative Syndrome Scale (PANSS)10 total scores at weeks 4 (primary efficacy endpoint), 9, and 25.9 The 3 most common adverse events (AEs) were injection site pain, increased weight, and akathisia.9
Because this was the first randomized clinical trial using a 2-month LAI formulation of aripiprazole (ie, AL 1064 mg) and was likewise the first in which the 1-day initiation regimen (ie, ALNCD plus 30-mg oral aripiprazole) was used, associated safety and tolerability data are of particular interest. A post hoc analysis was conducted to examine whether the extended dosing interval or the 1-day initiation regimen was associated with safety concerns not previously observed using monthly AL regimens initiated with 21 days of oral aripiprazole supplementation. The objective was to characterize the safety and tolerability of AL 1064 mg every 2 months (using PP 156 mg monthly as an active control) based on AEs of interest observed after inpatient initiation with the 1-day regimen or emerging during subsequent outpatient maintenance treatment of schizophrenia over 25 weeks. This analysis expands on the previously published ALPINE findings, focusing on the occurrence, timing, and severity of AEs associated with antipsychotic treatment.11–13
METHODS
Study Design and Patients
This study (ClinicalTrials.gov identifier: NCT03345979) was conducted between November 2017 and March 2019 and was approved by site-specific independent ethics committees/institutional review boards. Written informed consent was obtained from each participant.
Adults (aged 18–65 years) with schizophrenia according to Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition,14 experiencing an acute exacerbation requiring hospitalization were enrolled. Additional key enrollment criteria have been published.9 Imminent risk of suicide or a history of hypersensitivity or intolerance to aripiprazole, risperidone, or paliperidone were exclusionary.
Patients without prior exposure to aripiprazole and/or paliperidone/risperidone received test doses to assess tolerability during a ≤ 7-day screening period. Patients were randomized to double-blind treatment while hospitalized, discharged after 2 weeks if clinically stable, and continued outpatient treatment for the remainder of the 25-week study (Supplementary Figure 1).
Treatment
AL treatment was initiated on day 1 with an intramuscular (IM; gluteal) injection of ALNCD (675 mg) plus 30-mg oral aripiprazole. The first IM AL 1064-mg dose was administered on day 8 to parallel the requirements for PP initiation. AL 1064 mg was administered every 8 weeks thereafter from day 8. Treatment with PP was initiated with a 234-mg IM (deltoid) injection on day 1 followed by a 156-mg IM (deltoid) injection on day 8 and every 4 weeks (gluteal) thereafter. Because the initial PP doses were administered via deltoid injections, AL-treated patients received placebo deltoid injections on days 1 and 8 and placebo gluteal injections at weeks 5, 13, and 21 to maintain the blind. The PP group received an oral placebo tablet on day 1 and placebo gluteal injections on days 1 and 8 (Supplementary Figure 1).
Assessments
Specific AEs of interest included injection site reactions (ISRs; prespecified safety endpoint) and other events that could compromise continuous LAI antipsychotic pharmacotherapy: motor AEs, sedation, and hypotension. Weight gain, plasma prolactin, and suicidal ideation/behavior were also assessed.
Injection sites and surrounding areas were assessed 2–6 hours after injection, and potential ISRs were classified based on observed findings and subjective reports. Motor AEs, including dyskinesia, dystonia, drug-induced parkinsonism, and akathisia, were identified using standardized Medical Dictionary for Regulatory Activities (MedDRA) queries. Sedation AEs included sedation, somnolence, hypersomnia, and lethargy. Hypotension AEs included hypotension, orthostatic blood pressure, orthostatic hypotension, presyncope, syncope, dizziness, and tachycardia. Suicidal ideation or behavior AEs were assessed using the standardized MedDRA query suicide/self-injury. Because AEs were categorized differently to better present events of interest in this analysis, rates for some AEs reported here differ from those reported in the primary publication.
Assessments also included rating scales (Supplementary Table 1). Akathisia was defined as a global score ≥ 2 on the Barnes Akathisia Rating Scale (BARS).15 Parkinsonism was defined as total score > 3 on the Simpson-Angus Scale (SAS).16 Dyskinesia was defined as a score ≥ 3 on any of the first 7 items or ≥ 2 on 2 or more of these items on the Abnormal Involuntary Movement Scale (AIMS).17 An Epworth Sleepiness Scale (ESS)18 score ≥ 11 indicated excessive daytime sleepiness.19 Suicidal ideation or behavior was monitored using the Columbia-Suicide Severity Rating Scale (C-SSRS).20
Prolactin concentrations and body weight were determined at scheduled assessments (Supplementary Table 1). Change from baseline to last prolactin assessment and clinically significant prolactin elevations (> 1 ✕ upper limit of normal [ULN], > 2 ✕ ULN, > 3 ✕ ULN; > 100 ng/mL [males], and > 150 ng/mL [females]) were summarized by sex and treatment. Clinically significant increases in body weight (≥ 7% from baseline) were summarized by treatment.
Statistical Analyses
All patients who received ≥ 1 dose of study medication were included in this analysis. The study was not powered for between-group comparisons; results were summarized separately for AL and PP groups, with no formal statistical testing for comparing the two treatment arms. Descriptive statistics were used to report time to first occurrence of motor AEs and ISRs.
RESULTS
Patients
Of 200 patients randomized (AL, n = 99; PP, n = 101), 99 completed the study (AL, n = 56 [57%]; PP, n = 43 [43%]; Supplementary Table 2, Supplementary Figure 2). Mean (SD) age was similar between groups (AL, 43.5 [9.67] years; PP, 43.4 [10.83] years); ~75% of patients in each group were male. Mean (SD) baseline PANSS total scores were 94.1 (9.04) and 94.6 (8.41) for the AL and PP groups, respectively.
Aripiprazole Lauroxil
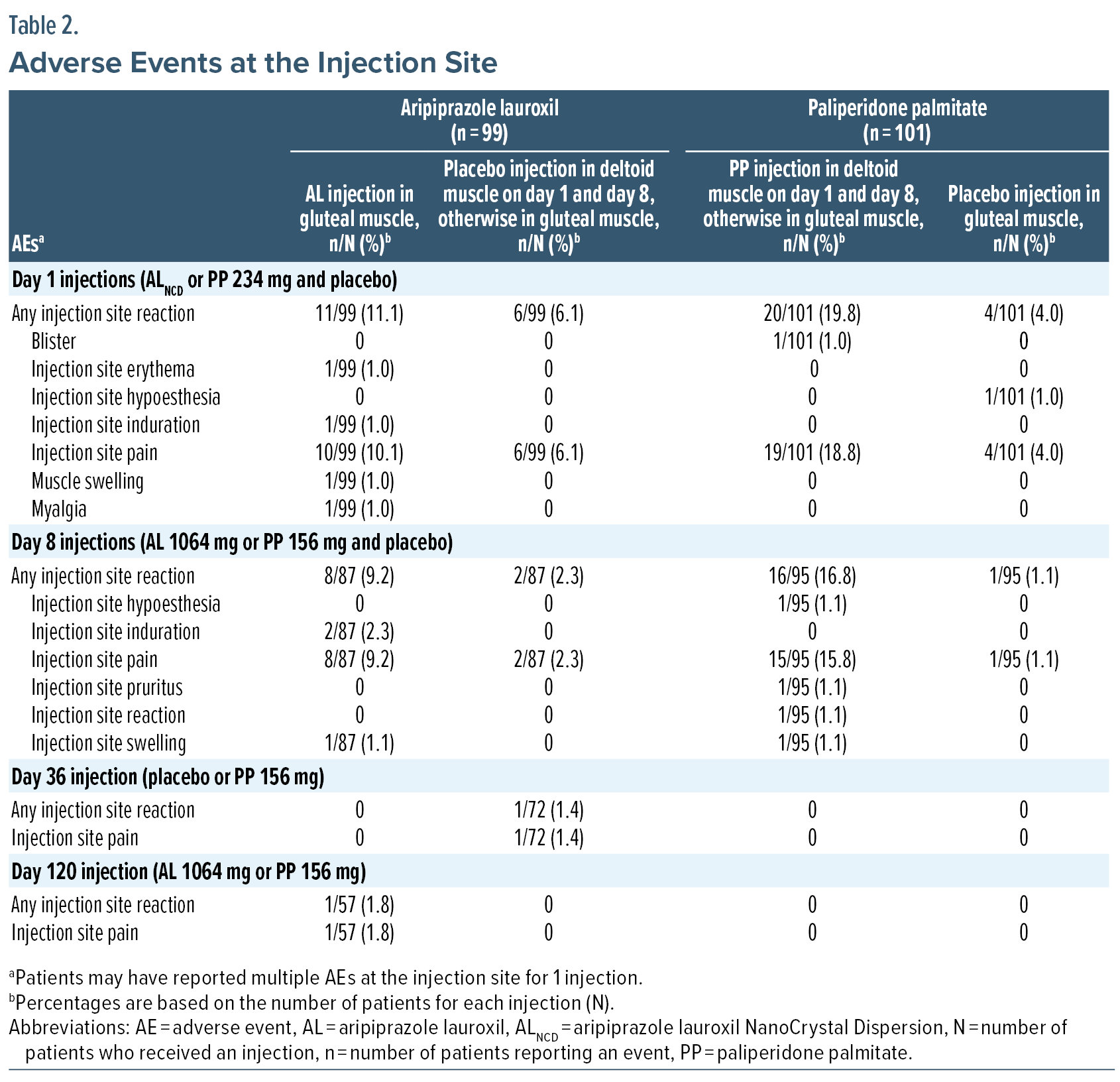
Injection site reactions. Following AL or placebo injections, ISRs were reported in 18/99 (18.2%) patients and were mild or moderately severe (Table 1); ISRs were predominantly associated with day 1 (ALNCD 675 mg) or day 8 (AL 1064 mg) injections (Table 2, Figure 1A). Injection site pain was the most frequent ISR (n = 17/99; 17.2%). Median time to first ISR was 1 day (mean, 5.1 days; range, 1–36 days). Two patients discontinued because of injection site pain: 1 because of pain at the deltoid injection site on day 1 (placebo) and 1 because of pain at the gluteal site on day 2 (ALNCD).
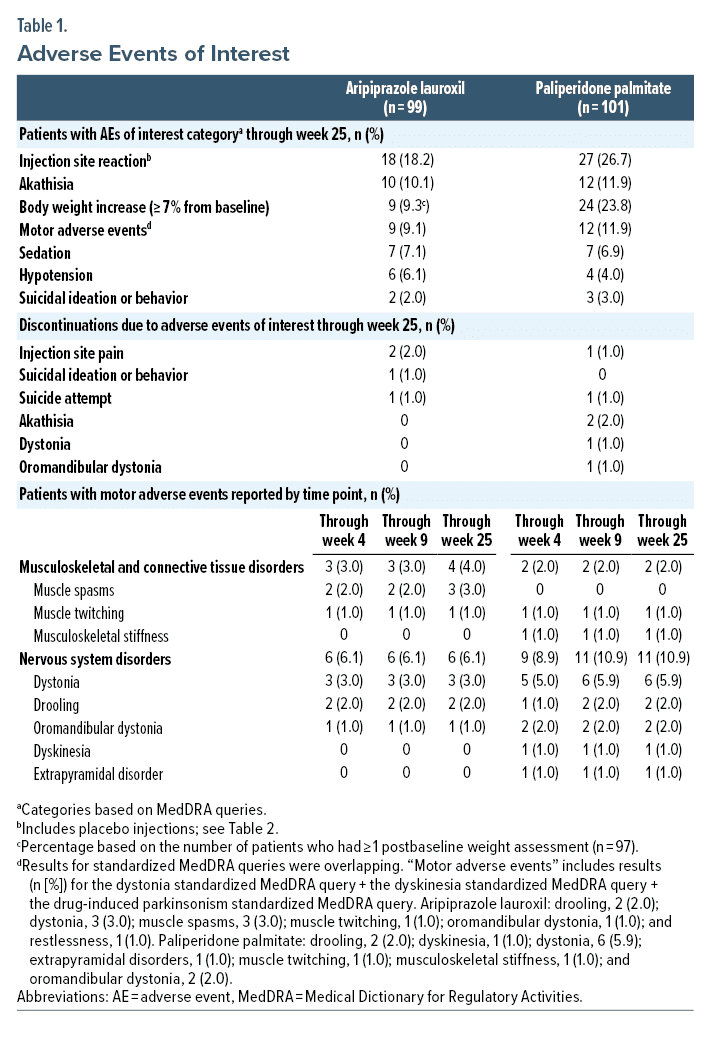
Motor adverse events, excluding akathisia. Nine of 99 (9.1%) AL-treated patients experienced motor AEs (dystonia, dyskinesia, and drug-induced parkinsonism; Table 1); in 8, motor AEs occurred within the first 4 weeks of treatment (Table 1). The remaining patient experienced a new event of mild muscle spasms after week 4. Median time to first motor AE event was 8 days (mean, 17.2 days; range, 1–101 days). All were mild or moderate in severity; none prompted discontinuation.
One patient met AIMS score criteria for dyskinesia and also experienced a mild AE of drooling (day 28). Four patients (4.2%) met the SAS criterion for parkinsonism; a corresponding AE was reported in only 1 patient (drooling).
Akathisia. Ten (10.1%) AL-treated patients experienced akathisia (preferred terms: akathisia [9 patients] and restlessness [1 patient]); no patients experienced > 1 episode. Median time to onset of akathisia was 3.5 days (mean, 6.5 days; range, 2–15 days). Most episodes were mild; 1 was moderate in severity. None led to discontinuation.
Akathisia defined by BARS global score occurred in 6/96 patients (6.3%). Four of those 6 also reported AEs of akathisia (3 mild, 1 moderate).
Sedation and daytime sleepiness. Seven of 99 (7.1%) AL-treated patients experienced AEs of sedation (Table 1); all were mild. In 5 of the 7 patients, sedation-related events occurred during the first 4 study weeks and resolved during the treatment period. Of the 2 patients with unresolved events, 1 discontinued because of worsening/exacerbation of schizophrenia symptoms. No sedation-related treatment discontinuations occurred. A total of 60/99 (60.6%) patients received concomitant benzodiazepines during the study; no AEs of sedation were reported in these patients.
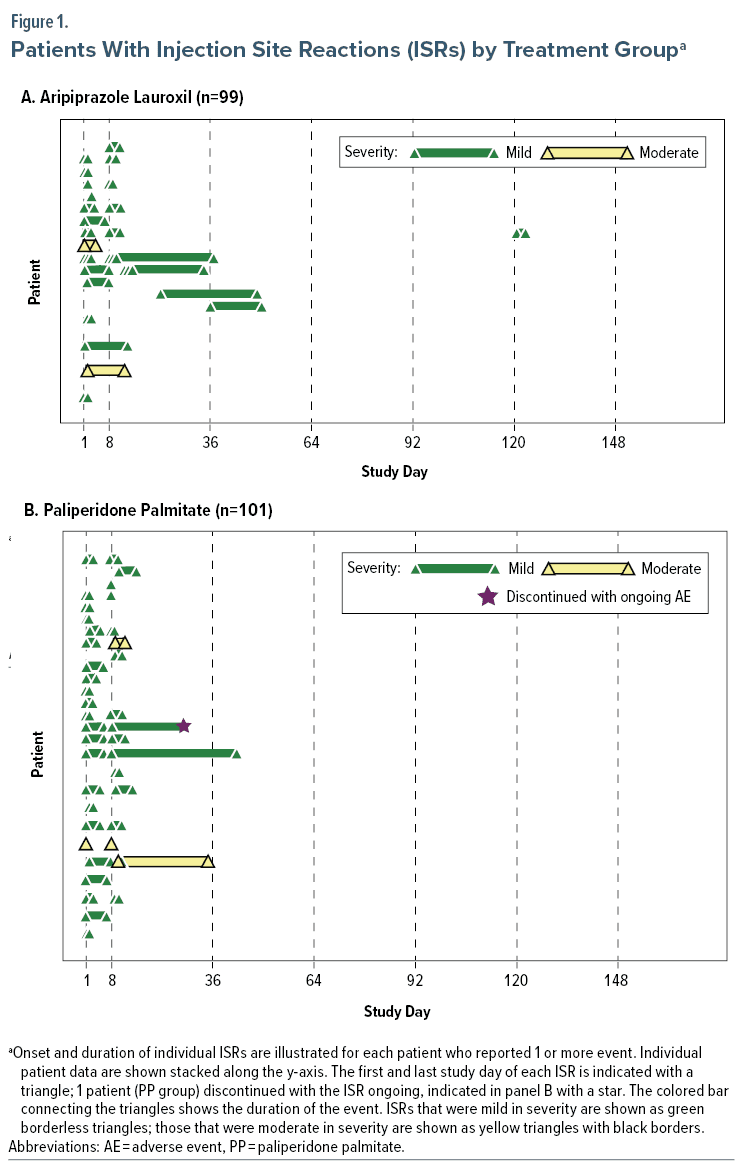
Mean (SD) ESS scores remained relatively stable from week 2 (7.3 [5.43]) through week 25 (7.9 [6.94]). Excessive daytime sleepiness (ESS ≥ 11) was observed in 29.8%–34.7% of patients across assessments (Figure 2A).
Hypotension. Hypotension-related AEs were reported in 6/99 (6.1%) AL-treated patients; 5 events were mild, and 1 was moderate. Most events were transient, and no patient discontinued treatment. A total of 27/99 (27.3%) patients received concomitant antihypertensive medications; none reported hypotension-related AEs.
Prolactin. Median change from baseline in prolactin concentration at last assessment was −4.60 ng/mL for males and −3.55 ng/mL for females (individual values over time are presented in Supplementary Figure 3). None of 70 males with postbaseline assessments had clinically significant concentrations > 1 ✕ ULN or > 100 ng/mL. One (4.5%) of 22 females with postbaseline assessments had concentrations > 1 ✕ ULN; none had concentrations > 2 ✕ ULN or > 150 ng/mL.
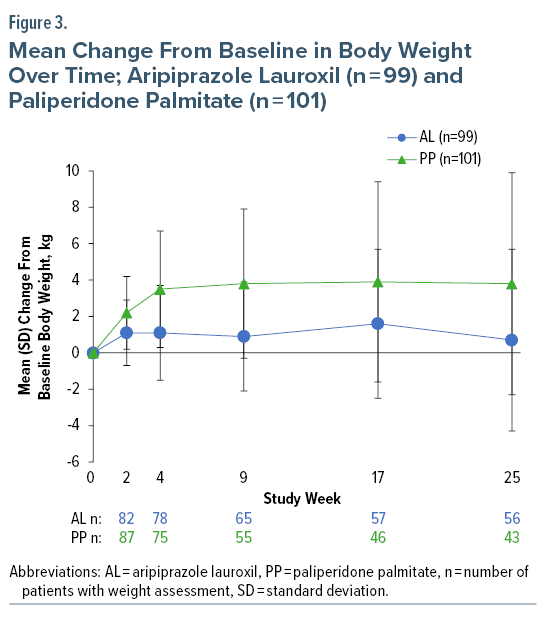
Body weight. Weight gain of ≥ 7% from baseline was observed at last assessment in 9/97 (9.3%) AL-treated patients; 5 were of normal weight at baseline, 3 were overweight, and 1 was obese. Weight loss of ≥ 7% from baseline was observed in 6/97 (6.2%) patients; 2 were of normal weight at baseline, 2 were overweight, and 2 were obese. Most weight change with AL occurred by week 2, with little mean change between weeks 2 and 25 (Figure 3).
Suicidal ideation/behavior. Suicidal ideation/behavior was reported in 2/99 (2.0%) AL-treated patients, all moderate in severity. One patient experienced 2 suicidal ideation events, the second leading to discontinuation (day 54). Both events were considered likely unrelated to treatment. One patient experienced nonspecific suicidal thoughts (day 122) and attempted suicide (entered oncoming traffic in response to auditory hallucinations) on day 134. The patient was hospitalized and discontinued from the study on day 147 because of suicide attempt and worsening/exacerbation of schizophrenia (each resolved on day 147), both were considered serious AEs and possibly treatment related. Suicidal ideation/behavior, assessed using the C-SSRS, was reported in 3 (3.0%) patients, including those reported as AEs; 1 (1.0%) patient attempted suicide.
Paliperidone Palmitate
Injection site reactions. Following PP or placebo injections, ISRs were reported in 27/101 (26.7%) patients and were mild or moderately severe (Table 1); injection site pain was the most frequently reported ISR (25 [24.8%]). All ISRs were observed following day 1 or day 8 injections, most after deltoid muscle administration (PP) (Table 2, Figure 1B). Median time to ISR onset was 1 day (mean, 2.3 days; range, 1–10 days). One patient discontinued because of moderate deltoid muscle injection site pain following the day 8 injection (PP).
Motor adverse events, excluding akathisia. Twelve of 101 (11.9%) PP-treated patients had motor AEs (dystonia, dyskinesia, and drug-induced parkinsonism; Table 1). Three patients had 2 motor AEs each. Most occurred during the first 4 weeks of treatment (Table 1), with 1 additional report of dystonia and drooling thereafter. Median time to a first motor AE was 13 days (mean, 16.2 days; range, 2–50 days). All motor AEs were mild or moderate in severity. Two (4.0%) patients discontinued because of dystonia.
Dyskinesia, defined by AIMS score, occurred in 1 patient, who also experienced an AE of mild oromandibular dystonia. Parkinsonism, defined by the SAS, occurred in 3 patients (3.0%), including the patient with AIMS-defined dyskinesia; the other 2 reported no motor AEs.
Akathisia. Twelve PP-treated patients (11.9%) experienced akathisia: 10 with single episodes and 2 patients with > 1 episode. All initial episodes occurred within the first 4 weeks. Median time to first akathisia experience was 7.5 days (mean, 8.5 days; range, 1–22 days). Most events were mild in severity; 2 patients experienced moderate akathisia (1 discontinuation), and 1 patient experienced severe akathisia resulting in discontinuation, with moderate akathisia reported during safety follow-up. Both patients who discontinued because of akathisia experienced the AE within the first 4 weeks of treatment; those patients discontinued on days 57 and 78, respectively.
Akathisia, defined by BARS score, occurred in 9/99 (9.1%) patients with ≥ 1 postbaseline assessment; of these, 7 also had akathisia AEs (2 moderate, 1 severe in severity).
Sedation and daytime sleepiness. Seven of 101 (6.9%) patients experienced sedation (Table 1). One event was moderately severe, and all others were mild; no patients discontinued. A total of 55/99 (54.5%) patients received benzodiazepines as concomitant medication during the study; 2 of these patients had sedation AEs.
Mean ESS scores remained relatively stable from week 2 (8.0 [4.93]) through week 25 (7.2 [6.26]); 32.1%–37.3% of patients had excessive daytime sleepiness (ESS ≥ 11) across assessments (Figure 2B).
Hypotension. Hypotension AEs were reported in 4 (4.0%) patients in the PP group (Table 1); all were mild or moderate in severity. One patient discontinued treatment because of dizziness. A total of 22/101 (21.8%) patients received concomitant antihypertensive medications; none had AEs related to hypotension.
Prolactin. Median change from baseline in prolactin concentration at last assessment was 21.2 ng/mL in males and 80.4 ng/mL in females (Supplementary Figure 3). Among assessed males (n = 73), prolactin concentrations were > 1 ✕ ULN in 59 (80.8%), > 2 ✕ ULN in 28 (38.4%), and > 3 ✕ ULN in 10 (13.7%); none had concentrations > 100 ng/mL. Among assessed females (n = 25), prolactin concentrations were > 1 ✕ ULN in 24 (96%), > 2 ✕ ULN in 22 (88.0%), and > 3 ✕ ULN in 13 (52.0%); 2 (8.0%) had concentrations > 150 ng/mL.
Body weight. Weight increases ≥ 7% from baseline occurred in 24/101 (23.8%) PP-treated patients; 11 were normal weight at baseline, and 13 were overweight (n = 9) or obese (n = 4). One patient (overweight at baseline) had a weight decrease ≥ 7% from baseline. The greatest mean change in body weight occurred by week 4, with little change thereafter (Figure 3).
Suicidal ideation/behavior. Suicidal ideation/behavior events were reported in 3/101 (3.0%) PP-treated patients (Table 1). Two patients experienced mild suicidal ideation on day 7 (likely unrelated to study drug) and day 35 (possibly related). Neither event resulted in discontinuation. A third patient experienced moderate suicidal ideation on day 35, attempted suicide (intentional overdose using zolpidem and lorazepam) on day 96, and was discontinued on day 134; overdose and suicide attempt were judged as serious events unrelated to the study drug. The C-SSRS captured suicidal ideation/behavior in 7 (6.9%) patients and a suicide attempt in 1 patient, as described above.
DISCUSSION
Based on occurrence and timing of AEs in this post hoc analysis of randomized, active-controlled study data, the safety profiles of AL and PP were consistent with those reported in previous studies.12,21–28 For both, most AEs of interest had their onset within the first 4 weeks of treatment initiation. No new early- or late-emerging safety concerns were observed through 25 weeks of treatment with AL 1064 mg every 2 months following initiation using ALNCD plus 30-mg oral aripiprazole. The low rates of ≥ 7% weight gain and single case of elevated prolactin (> 1✕ ULN) observed with AL 1064 mg every 2 months were consistent with published findings for monthly AL regimens initiated using 21-day oral aripiprazole supplementation.12,22
Despite the potential benefits of LAI antipsychotics, including predictable and consistent plasma drug levels,29 they are underused for the treatment of schizophrenia.30 Both patients and clinicians cite apprehension about needles as a top reason for rejecting LAI formulations.31–33 The tolerability of LAIs is affected by ISRs, and injection site pain is a common concern for patients.31–34 In this analysis, ISRs were mild or moderately severe and mainly associated with day 1 or day 8 injections. The most frequent ISR, injection site pain, was uncommon with later injections, consistent with published observations that pain decreases over time with LAI antipsychotic use.23,35–37 Placebo injections used to maintain blinding may have increased ISR rates over what might be expected with AL or PP injections alone, particularly for patients assigned to AL, who received a greater number of placebo injections than AL injections. The use of formulations with extended injection intervals can reduce the number of injections during treatment with LAI antipsychotics, potentially mitigating fear of needles as a barrier to their use.31,34
Motor AEs and akathisia are commonly associated with antipsychotic treatment,11,12,38–40 and an increased risk has been observed with higher-dose treatment for some antipsychotics.12,40,41 In the current analysis, motor AEs were experienced by approximately 10% of patients receiving AL and PP and generally occurred early, consistent with previous studies.12,30,38,39 Among patients treated with AL, motor AEs were predominantly mild or moderate in severity, and the 1 new motor event reported after week 4 was mild. No motor AEs or AEs of akathisia were severe or led to discontinuation of AL treatment in this study. Not all patients meeting rating scale criteria for dystonia, parkinsonism, or akathisia reported related AEs; however, the overall percentages meeting criteria for these movement disorders were limited, and these relationships were not formally assessed. The 1064-mg dosage strength of the AL every-2-month regimen is higher than those used in monthly AL regimens.7 However, pharmacokinetic analysis indicates that plasma aripiprazole exposure achieved after administration of AL 1064 mg every 2 months is within the range observed for the 441-mg and 882-mg AL monthly regimens,42 suggesting that no increased risk of motor AEs would be expected with the every-2-month regimen. Treatment with AL 1064 mg every 2 months initiated using ALNCD plus 30-mg oral aripiprazole was not associated with severe motor AEs or akathisia during initiation, or worsening of events afterward.
This post hoc analysis had several limitations. The study was not powered for direct comparisons between AL and PP; the PP group provided an active control with known efficacy.9 The absence of a placebo group limits the interpretability of results, and ISR frequencies may be overestimated because placebo injections were administered to maintain blinding. The limited patient numbers and fixed-dose design may not extrapolate to all patients with schizophrenia receiving AL. Although the highest PP dose available (234 mg) was not used for maintenance treatment, the intermediate dose chosen (156 mg monthly; equivalent to oral paliperidone extended release 9 mg/d)43 was consistent with the use of an intermediate dose of AL, 1064 mg every 2 months (equivalent to oral aripiprazole 15 mg/d).7 In this study, the initiation regimen of ALNCD plus 30-mg oral aripiprazole on day 1 was separated by 1 week from the first maintenance dose of AL 1064 mg to maintain the study blinding; however, the different pharmacokinetic characteristics of each aripiprazole formulation allow for an initiation strategy where the ALNCD and AL injections could be given in a single day, as described in product labeling.6,7
Conclusions
Patients prescribed LAI antipsychotics may prefer extended dosing intervals to reduce the numbers of injections received over time.31 In this post hoc analysis, the AE profile for every-2-month AL maintenance following initiation using ALNCD and 30-mg oral aripiprazole was consistent with the established safety of AL monthly dosing regimens started with 21 days of oral aripiprazole supplementation.12,21–23 For both AL and PP, the most common AEs (ISRs, motor AEs, and ≥ 7% weight gain) generally occurred early in treatment and were infrequent beyond week 4. No new safety concerns were observed after inpatient initiation of AL or PP, and none emerged over time after transition to outpatient care in this post hoc analysis of 25-week ALPINE study data. These results support the safety and tolerability of AL 1064 mg administered every 2 months following initiation using ALNCD plus 30-mg oral aripiprazole.
Article Information
Published Online: February 28, 2024. https://doi.org/10.4088/JCP.23m15095
© 2024 Physicians Postgraduate Press, Inc.
Submitted: September 11, 2023; accepted January 18, 2024.
To Cite: Citrome L, Yagoda S, Bidollari I, et al. Safety and tolerability of starting aripiprazole lauroxil with aripiprazole lauroxil NanoCrystal Dispersion in 1 day followed by aripiprazole lauroxil every 2 months using paliperidone palmitate monthly as an active control in patients with schizophrenia: a post hoc analysis of a randomized controlled trial. J Clin Psychiatry. 2024;85(1):23m15095.
Author Affiliations: New York Medical College, Valhalla, New York (Citrome); Alkermes, Inc., Waltham, Massachusetts (Yagoda, Bidollari, Wang). Drs Bidollari and Wang were employees of Alkermes, Inc., at the time of the analysis.
Corresponding Author: Leslie Citrome, MD, MPH, New York Medical College, 40 Sunshine Cottage Rd, Valhalla, NY 10595 ([email protected]).
Notice of correction 10/20/2025: In Table 2, in the “AL injection in gluteal muscle” column, values for the following adverse events have been corrected: in the Day 1 injections section: injection site induration, muscle swelling, and myalgia; in the Day 8 injections section: injection site swelling.
Author Contributions: Study design: (Yagoda); collection and assembly of data: (Yagoda); data analysis: (all authors); data interpretation: (all authors); manuscript preparation: (all authors); manuscript review and revisions: (all authors).
Relevant Financial Relationships: Dr Citrome has served as consultant to AbbVie/Allergan, Acadia, Adamas, Alkermes (including during conduct of this study), Angelini, Astellas, Avanir, Axsome, BioXcel, Boehringer Ingelheim, Cadent Therapeutics, Cerevel, Clinilabs, COMPASS, Eisai, Enteris BioPharma, HLS Therapeutics, Idorsia, Impel, INmune Bio, Intra-Cellular Therapies, Janssen, Karuna, Lundbeck, Lyndra, MedAvante-ProPhase, Marvin, Merck, Mitsubishi-Tanabe Pharma, Neurelis, Neurocrine, Novartis, Noven, Otsuka, Ovid, Praxis, Recordati, Relmada, Reviva, Sage, Sunovion, Supernus, Teva, University of Arizona, Vanda, and one-off ad hoc consulting for individuals/entities conducting marketing, commercial, or scientific scoping research; received speaker fees from AbbVie/Allergan, Acadia, Alkermes, Angelini, Axsome, BioXcel, Eisai, Idorsia, Intra-Cellular Therapies, Janssen, Lundbeck, Neurocrine, Noven, Otsuka, Recordati, Sage, Sunovion, Takeda, Teva, and CME activities organized by medical education companies such as Medscape, NACCME, NEI, Vindico, and universities and professional organizations/societies; received fees/royalties/publishing income for work with Elsevier (Topic Editor, Psychiatry, Clinical Therapeutics), Springer Healthcare (book), Taylor & Francis (Editor-in-Chief, Current Medical Research and Opinion, 2022-date), UpToDate (reviewer), and Wiley (Editor-in-Chief, International Journal of Clinical Practice, through end 2019); owns a small number of shares of common stock (purchased >10 years ago) in Bristol Myers Squibb, Eli Lilly, J&J, Merck, and Pfizer; and has stock options with Reviva. Drs Yagoda, Bidollari, and Wang are or were employees of Alkermes, Inc., at the time this study was conducted and may own stock/options in the company.
Funding/Support: This study was sponsored by Alkermes, Inc. (Waltham, MA).
Role of the Sponsor: Employees of Alkermes, Inc., were involved in the study design and data analyses. The authors interpreted the evidence and made the decision to submit the manuscript for publication.
Previous Presentation: Presented at Psych Congress 2020, September 10–13, 2020.
Acknowledgments: Medical writing and editorial support were provided by Kathleen M. Dorries, PhD; Kalpana Vijayan, PhD; and John H. Simmons, MD, of Peloton Advantage, LLC (Parsippany, NJ, USA), an OPEN Health company, and funded by Alkermes, Inc.
Data Sharing Statement: The data used in the preparation of this manuscript are proprietary to Alkermes, Inc. Alkermes, Inc., is committed to public sharing of data in accordance with applicable regulations and laws, and requests can be submitted to the corresponding author.
ORCID: Leslie Citrome: 0000-0002-6098-9266.
Supplementary Material: Available at Psychiatrist.com.
Clinical Points
- This post hoc analysis evaluated adverse events of interest after inpatient initiation and subsequent outpatient maintenance treatment with aripiprazole lauroxil (AL) 1064 mg every 2 months or paliperidone palmitate 156 mg monthly (as active control) over a period of 25 weeks.
- AL was initiated using one intramuscular injection of the NanoCrystal Dispersion formulation of AL (ALNCD) plus one 30-mg dose of oral aripiprazole, followed by AL 1064 mg 1 week later; PP was initiated using 234-mg and 156-mg intramuscular injections 1 week apart. Evaluations continued during outpatient treatment through week 25 of treatment.
- Treatment of patients with acutely exacerbated schizophrenia with AL or PP was well tolerated, consistent with the respective known safety profiles of AL and PP.
References (43)

- American Psychiatric Association. The American Psychiatric Association Practice Guideline for the Treatment of Patients With Schizophrenia. American Psychiatric Association; 2021.
- Correll CU, Martin A, Patel C, et al. Systematic literature review of schizophrenia clinical practice guidelines on acute and maintenance management with antipsychotics. Schizophrenia (Heidelb). 2022;8(1):5. PubMed CrossRef
- Citrome L. Long-acting injectable antipsychotics update: lengthening the dosing interval and expanding the diagnostic indications. Expert Rev Neurother. 2017;17(10):1029–1043. PubMed CrossRef
- Faden J, Citrome L. How would you like to take your medicine 2 times a year? Paliperidone palmitate every 6 months for the maintenance treatment of schizophrenia. Clin Ther. 2022;44(4):476–479. PubMed CrossRef
- Ceskova E, Silhan P. Novel treatment options in depression and psychosis. Neuropsychiatr Dis Treat. 2018;14:741–747. PubMed CrossRef
- Jain R, Meyer J, Wehr A, et al. Size matters: the importance of particle size in a newly developed injectable formulation for the treatment of schizophrenia. CNS Spectr. 2020;25(3):323–330. PubMed CrossRef
- ARISTADA INITIO. Package insert. Alkermes, Inc.; 2021.
- Weiden PJ, Claxton A, Kunovac J, et al. Efficacy and safety of a 2-month formulation of aripiprazole lauroxil with 1-day initiation in patients hospitalized for acute schizophrenia transitioned to outpatient care: phase 3, randomized, double-blind, active control ALPINE study. J Clin Psychiatry. 2020;81(3):19m13207.
- Kay SR, Fiszbein A, Opler LA. The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13(2):261–276. PubMed CrossRef
- Gentile S. Adverse effects associated with second-generation antipsychotic long-acting injection treatment: a comprehensive systematic review. Pharmacotherapy. 2013;33(10):1087–1106. PubMed CrossRef
- Nasrallah HA, Aquila R, Du Y, et al. Long-term safety and tolerability of aripiprazole lauroxil in patients with schizophrenia. CNS Spectr. 2019;24(4):395–403. PubMed CrossRef
- Citrome L. Long-acting injectable antipsychotics: what, when, and how. CNS Spectr. 2021;26(2):118–129. PubMed CrossRef
- American Psychiatric Association. Schizophrenia Spectrum and Other Psychiatric Disorders. In: Diagnostic and Statistical Manual of Mental Disorders. 5th ed. American Psychiatric Association; 2013.
- Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154(5):672–676. PubMed CrossRef
- Simpson GM, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand. 1970;45(S212):11–19. PubMed CrossRef
- Guy W. Abnormal Involuntary Movement Scale (AIMS). ECDEU Assessment Manual for Psychopharmacology. US Department of Health, Education, and Welfare, National Institute of Mental Health; 1976:534–537.
- Johns MW. A new method for measuring daytime sleepiness: the Epworth Sleepiness Scale. Sleep. 1991;14(6):540–545. PubMed CrossRef
- Johns M, Hocking B. Daytime sleepiness and sleep habits of Australian workers. Sleep. 1997;20(10):844–849. PubMed CrossRef
- Johns M, Hocking B. Daytime sleepiness and sleep habits of Australian workers. Sleep. 1997;20(10):844–849. PubMed CrossRef
- Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266–1277. PubMed CrossRef
- Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085–1090. PubMed CrossRef
- Weiden PJ, Du Y, Liu CC, et al. Switching stable patients with schizophrenia from their oral antipsychotics to aripiprazole lauroxil: a post hoc safety analysis of the initial 12-week crossover period. CNS Spectr. 2019;24(4):419–425. PubMed CrossRef
- Lauriello J, Claxton A, Du Y, Weiden PJ. Beyond 52-week long-term safety: long-term outcomes of aripiprazole lauroxil for patients with schizophrenia continuing in an extension study. J Clin Psychiatry. 2020;81(5):19m12835.
- Kramer M, Litman R, Hough D, et al. Paliperidone palmitate, a potential long-acting treatment for patients with schizophrenia. Results of a randomized, double-blind, placebo-controlled efficacy and safety study. Int J Neuropsychopharmacol. 2010;13(5):635–647. PubMed CrossRef
- Gopal S, Hough DW, Xu H, et al. Efficacy and safety of paliperidone palmitate in adult patients with acutely symptomatic schizophrenia: a randomized, double-blind, placebo-controlled, dose-response study. Int Clin Psychopharmacol. 2010;25(5):247–256. PubMed CrossRef
- Li H, Rui Q, Ning X, et al. A comparative study of paliperidone palmitate and risperidone long-acting injectable therapy in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(4):1002–1008. PubMed CrossRef
- Pandina G, Lane R, Gopal S, et al. A double-blind study of paliperidone palmitate and risperidone long-acting injectable in adults with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(1):218–226. PubMed CrossRef
- Gopal S, Vijapurkar U, Lim P, et al. A 52-week open-label study of the safety and tolerability of paliperidone palmitate in patients with schizophrenia. J Psychopharmacol. 2011;25(5):685–697. PubMed CrossRef
- Milz R, Benson C, Knight K, et al. The effect of longer dosing intervals for long-acting injectable antipsychotics on outcomes in schizophrenia. Neuropsychiatr Dis Treat. 2023;19:531–545. PubMed CrossRef
- Correll CU, Citrome L, Haddad PM, et al. The use of long-acting injectable antipsychotics in schizophrenia: evaluating the evidence. J Clin Psychiatry. 2016;77(suppl 3):1–24. PubMed CrossRef
- Blackwood C, Sanga P, Nuamah I, et al. Patients’ preference for long-acting injectable versus oral antipsychotics in schizophrenia: results from the patient-reported medication preference questionnaire. Patient Prefer Adherence. 2020;14:1093–1102. PubMed CrossRef
- Potkin S, Bera R, Zubek D, et al. Patient and prescriber perspectives on long-acting injectable (LAI) antipsychotics and analysis of in-office discussion regarding LAI treatment for schizophrenia. BMC Psychiatry. 2013;13(1):261. PubMed CrossRef
- Cahling L, Berntsson A, Bröms G, et al. Perceptions and knowledge of antipsychotics among mental health professionals and patients. BJPsych Bull. 2017;41(5):254–259. PubMed CrossRef
- Parellada E, Bioque M. Barriers to the use of long-acting injectable antipsychotics in the management of schizophrenia. CNS Drugs. 2016;30(8):689–701. PubMed CrossRef
- Kern Sliwa J, Savitz A, Nuamah I, et al. An assessment of injection site reaction and injection site pain of 1-month and 3-month long-acting injectable formulations of paliperidone palmitate. Perspect Psychiatr Care. 2018;54(4):530–538. PubMed CrossRef
- Calabrese JR, Sanchez R, Jin N, et al. Efficacy and safety of aripiprazole once-monthly in the maintenance treatment of bipolar I disorder: a double-blind, placebo-controlled, 52-week randomized withdrawal study. J Clin Psychiatry. 2017;78(3):324–331. PubMed CrossRef
- Fleischhacker WW, Eerdekens M, Karcher K, et al. Treatment of schizophrenia with long-acting injectable risperidone: a 12-month open-label trial of the first long-acting second-generation antipsychotic. J Clin Psychiatry. 2003;64(10):1250–1257. PubMed CrossRef
- Gopal S, Liu Y, Alphs L, et al. Incidence and time course of extrapyramidal symptoms with oral and long-acting injectable paliperidone: a posthoc pooled analysis of seven randomized controlled studies. Neuropsychiatr Dis Treat. 2013;9:1381–1392. PubMed CrossRef
- Mathews M, Nuamah I, Savitz AJ, et al. Time to onset and time to resolution of extrapyramidal symptoms in patients with exacerbated schizophrenia treated with 3-monthly vs once-monthly paliperidone palmitate. Neuropsychiatr Dis Treat. 2018;14:2807–2816. PubMed CrossRef
- Chow CL, Kadouh NK, Bostwick JR, et al. Akathisia and newer second-generation antipsychotic drugs: a review of current evidence. Pharmacotherapy. 2020;40(6):565–574. PubMed CrossRef
- Shirzadi AA, Ghaemi SN. Side effects of atypical antipsychotics: extrapyramidal symptoms and the metabolic syndrome. Harv Rev Psychiatry. 2006;14(3):152–164. PubMed CrossRef
- Hard ML, Mills RJ, Sadler BM, et al. Pharmacokinetic profile of a 2-month dose regimen of aripiprazole lauroxil: a phase I study and a population pharmacokinetic model. CNS Drugs. 2017;31(7):617–624. PubMed CrossRef
- INVEGA SUSTENNA. Package insert. Janssen Pharmaceuticals; 2021.
This PDF is free for all visitors!





