ABSTRACT
Objective: Altered glutamatergic neurotransmission has been implicated in the pathogenesis of depression. This trial evaluated the efficacy and safety of AXS-05 (dextromethorphan-bupropion), an oral N-methyl-D-aspartate (NMDA) receptor antagonist and σ1 receptor agonist, in the treatment of major depressive disorder (MDD).
Methods: This double-blind, phase 3 trial, was conducted between June 2019 and December 2019. Patients with a DSM-5 diagnosis of MDD were randomized in a 1:1 ratio to receive dextromethorphan-bupropion (45 mg-105 mg tablet) or placebo, orally (once daily for days 1–3, twice daily thereafter) for 6 weeks. The primary endpoint was the change from baseline to week 6 in the Montgomery-Asberg Depression Rating Scale (MADRS) total score. Other efficacy endpoints and variables included MADRS changes from baseline at week 1 and 2, clinical remission (MADRS score ≤ 10), clinical response (≥ 50% reduction in MADRS score from baseline), clinician- and patient-rated global assessments, Quick Inventory of Depressive Symptomatology-Self-Rated, Sheehan Disability Scale, and quality of life measures.
Results: A total of 327 patients were randomized: 163 patients to dextromethorphan-bupropion and 164 patients to placebo. Mean baseline MADRS total scores were 33.6 and 33.2 in the dextromethorphan-bupropion and placebo groups, respectively. The least-squares mean change from baseline to week 6 in MADRS total score was −15.9 points in the dextromethorphan-bupropion group and −12.0 points in the placebo group (least-squares mean difference, −3.87; 95% confidence interval [CI], −1.39 to −6.36; P = .002). Dextromethorphan-bupropion was superior to placebo for MADRS improvement at all time points including week 1 (P = .007) and week 2 (P < .001). Remission was achieved by 39.5% of patients with dextromethorphan-bupropion versus 17.3% with placebo (treatment difference, 22.2; 95% CI, 11.7 to 32.7; P < .001), and clinical response by 54.0% versus 34.0%, respectively (treatment difference, 20.0%; 95% CI, 8.4%, 31.6%; P < .001), at week 6. Results for most secondary endpoints were significantly better with dextromethorphan-bupropion than with placebo at almost all time points (eg, CGI-S least-squares mean difference at week 6, −0.48; 95% CI, −0.48 to −0.79; P = .002). The most common adverse events in the dextromethorphan-bupropion group were dizziness, nausea, headache, somnolence, and dry mouth. Dextromethorphan-bupropion was not associated with psychotomimetic effects, weight gain, or increased sexual dysfunction.
Conclusions: In this phase 3 trial in patients with MDD, treatment with dextromethorphan-bupropion (AXS-05) resulted in significant improvements in depressive symptoms compared to placebo starting 1 week after treatment initiation and was generally well tolerated.
Trial Registration: ClinicalTrials.gov Identifier: NCT04019704
J Clin Psychiatry 2022;83(4):21m14345
To cite: Iosifescu DV, Jones A, O’Gorman C, et al. Efficacy and safety of AXS-05 (dextromethorphan-bupropion) in patients with major depressive disorder: a phase 3 randomized clinical trial (GEMINI). J Clin Psychiatry. 2022;83(4):21m14345.
To share: https://doi.org/10.4088/JCP.21m14345
© 2022 Physicians Postgraduate Press, Inc.
aNathan Kline Institute and New York University School of Medicine, New York, New York
bAxsome Therapeutics, Inc, New York, New York
cMassachusetts General Hospital Clinical Trials Network and Institute, Boston, Massachusetts
dDepartment of Psychiatry, Harvard Medical School, Boston, Massachusetts
eDr O’Gorman is no longer affiliated with Axsome Therapeutics.
*Corresponding author: Amanda Jones, PharmD, Axsome Therapeutics, 22 Cortland St, New York, NY 10007 ([email protected]).
Major depressive disorder (MDD) is a prevalent, disabling, chronic, biologically based disorder, which impairs social, occupational, and educational functioning.1 It is the leading cause of disability worldwide and is associated with increased suicide risk, morbidity, and mortality.2,3 Currently approved oral antidepressants work primarily via monoamine pathways.4 Partial or inadequate response is common with these agents, and they typically take several weeks to produce clinically meaningful effects.5 In the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial, about two thirds of depression patients failed to achieve remission with first-line treatment, and of those who experienced a clinical response, approximately 60% did so only at or after 8 weeks of treatment.6
Involvement of the glutamatergic system in the pathogenesis of depression is suggested by data from neuroimaging, cellular, and clinical studies. This evidence includes findings of abnormal glutamate levels in the cortex of depressed patients using magnetic resonance spectroscopy,7 observations of abnormal N-methyl-D-aspartate (NMDA) receptor expression and signaling in postmortem cortical preparations from depressed patients,8 and demonstration of antidepressant efficacy in studies with parenteral administration of the NMDA receptor antagonist ketamine.9,10
Dextromethorphan is an uncompetitive antagonist of the NMDA receptor (an ionotropic glutamate receptor)11 and a σ1 receptor agonist.12 Blockade of the NMDA receptor and agonism of the σ1 receptor modulate glutamate signaling in the central nervous system.13,14 The clinical utility of dextromethorphan has been limited in humans by its rapid and extensive metabolism through cytochrome P450 (CYP)2D6 yielding subtherapeutic plasma levels.15 A tablet (AXS-05) combining dextromethorphan and bupropion (hereafter, dextromethorphan-bupropion) has been formulated to increase the bioavailability and half-life of dextromethorphan and has been developed for the treatment of MDD. The bupropion component serves to increase plasma dextromethorphan concentrations by inhibiting its metabolism. Breakthrough Therapy designation was granted by the US Food and Drug Administration for dextromethorphan-bupropion for the treatment of MDD in March 2019. The objective of this phase 3 trial was to assess the efficacy and safety of dextromethorphan-bupropion compared to placebo in the treatment of patients with MDD.
METHODS
Trial Design and Oversight
The GEMINI (Glutamatergic and Monoaminergic Modulation in Depression) study was a phase 3, randomized, double-blind, placebo-controlled, 6-week trial conducted at 40 centers in the United States from June 2019 to December 2019. The trial was conducted in accordance with the International Council on Harmonization guidelines for Good Clinical Practice16 and the principles of the Declaration of Helsinki.17 The site investigators gathered the trial data, and the sponsor ensured that all persons administering rating scales were qualified and appropriately trained. All sites gained independent review board approval, and all patients provided written informed consent prior to participation.
Patient Population
Patients were men or women 18–65 years of age with a primary diagnosis of MDD, experiencing a major depressive episode of at least 4 weeks in duration, and having a Montgomery-Asberg Depression Rating Scale (MADRS)18 total score of 25 or higher, corresponding to moderate or greater severity, with higher scores indicating more severe depression. Patients were also required to have a score on the Clinician Global Impression-Severity (CGI-S)19 scale of 4 or higher (range, 1 to 7, with higher scores indicating greater severity of illness). The diagnosis of depression was established using the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5), criteria for MDD without psychotic features, based on the Structured Clinical Interview,20 which has been shown to diagnose MDD more conservatively than other structured interviews.21 As part of the screening process, an independent assessor confirmed the eligibility and symptom severity of each patient. The assessment consisted of a clinical review of all available documentation including complete medical history, and clinician- and patient-reported outcome measures.
Key exclusion criteria included bipolar disorder, psychotic disorder, panic disorder, obsessive-compulsive disorder, treatment-resistant depression (defined as ≥ 2 adequate failed antidepressant treatments in the current major depressive episode), alcohol/substance use disorder within the past year, clinically significant risk of suicide, and history of seizure disorder.
Trial Design and Procedures
Eligible patients were randomly assigned in a 1:1 ratio to receive either dextromethorphan-bupropion (45 mg-105 mg) or placebo orally for 6 weeks. Randomization was performed by a central interactive web response system. Study medication was provided by the trial sponsor and was identical in form and appearance; all investigators, patients, and study personnel involved in the study were blinded to study treatment. Patients received their assigned study medication once daily for 3 days, then twice daily thereafter. Study visits occurred at 1, 2, 3, 4, and 6 weeks after the baseline visit. A safety follow-up visit occurred at week 7, 1 week following the last dose of study medication. There were no formal discontinuation criteria; however, patients were free to withdraw consent for any reason and investigators were free to remove a patient from study for any safety-related reason. The dose of dextromethorphan-bupropion, titrated to twice daily, was selected based on the results of pharmacokinetic trials. The clinical trial was listed on ClinicalTrials.gov (NCT04019704).
Endpoints
The primary endpoint was the change from baseline to week 6 in the MADRS total score. The MADRS is a 10-item clinician-rated questionnaire ranging from 0 to 60, with higher scores representing more severe depression. The key secondary endpoints were the change from baseline in the MADRS total score at week 1; change from baseline in the MADRS total score at week 2; remission, defined as MADRS total score ≤ 10, at week 2; and clinical response, defined as ≥ 50% reduction in MADRS total score, at week 6.
Other secondary endpoints included the Clinician Global Impression-Improvement (CGI-I; scores range from 1 [very much improved] to 7 [very much worse])19; CGI-S (scores range from 1 [normal state] to 7 [among the most extremely ill])19; Patient Global Impression-Improvement (PGI-I; scores range from 1 [very much improved] to 7 [very much worse])19; Quick Inventory of Depressive Symptomatology-Self-Rated (QIDS-SR-16; scores range from 0 to 27 with higher scores representing more severe depression)22; Sheehan Disability Scale (SDS; scores range from 0 to 30 with higher scores indicating more severe disability)23; the Quality of Life Enjoyment and Satisfaction Questionnaire-Short Form (Q-LES-Q-SF; scores are based on the percentage of the maximum total score with higher percentages indicating greater satisfaction)24; and the MADRS-6 (a subscale of the 10-item MADRS evaluating the core symptoms of depression [apparent sadness, reported sadness, inner tension, lassitude, inability to feel, and pessimistic thought]).25,26
Safety was assessed based on the incidence of adverse events; changes in vital signs, clinical laboratory measurements, physical examinations, and electrocardiograms; assessment of suicidal ideation and behavior, with the use of the Columbia-Suicide Severity Rating Scale (C-SSRS); and assessment for withdrawal-related symptoms, using the Physician Withdrawal Checklist. Adverse events during the treatment period were defined as adverse events occurring from the time of administration of the first dose of dextromethorphan-bupropion or placebo until 7 days after the last dose.
Statistical Analysis
The safety analysis set included all patients who received at least 1 dose of study medication. Efficacy analyses were performed on the modified intent-to-treat population, which consisted of all patients who were randomized, received at least 1 dose of study medication, and had at least 1 post-baseline efficacy assessment. The primary efficacy variable, change from baseline in MADRS, was analyzed using a mixed model for repeated measures. This analysis of covariance mixed-effect model for repeated measures included treatment, week, and treatment-by-week interaction as factors, baseline value as a covariate, and patient as a random effect. All other change from baseline efficacy variables were analyzed using this method. Treatment effects and treatment differences at each time point were estimated using the least squares mean estimates. Efficacy variables related to percentages (eg, clinical response and remission rates, CGI-I, PGI-I) were performed on observed cases and analyzed via χ2 tests. Analyses were performed using SAS Version 9.4, and all hypothesis tests were conducted at a 2-sided α level of 0.05.
If results were found to be positive on the MADRS primary endpoint, then other analyses (response, remission) were to be performed on this variable to examine clinical relevance. To adjust for multiplicity, if the primary endpoint was established, then key secondary endpoints were examined with the use of hierarchical hypothesis tests in a pre-specified fixed-sequence procedure with the following order (we would stop after reaching a nonsignificant result): change in MADRS total score from baseline to week 2; percentage of patients achieving remission, defined as MADRS total score of ≤ 10, at week 2; change in MADRS total score from baseline to week 1; percentage of patients achieving clinical response, defined as ≥ 50% reduction in MADRS total score, at week 6; CGI-I at week 6; change in CGI-S from baseline to week 6; CGI-I at week 1; percentage of patients achieving remission at week 1; and change in SDS from baseline to week 6. For all other secondary endpoints, no adjustment for multiplicity was performed and nominal P values for these are presented.
There was no imputation of missing data for primary or secondary endpoints. Sensitivity analyses for the primary efficacy variable were performed based on the random replacement and tipping point methods.
A sample size of approximately 150 patients per treatment group would provide 90% power to detect a treatment difference on the primary efficacy variable of change in MADRS, at a 2-sided significance level of 0.05, assuming an effect size of 0.31.
RESULTS
Patient Characteristics
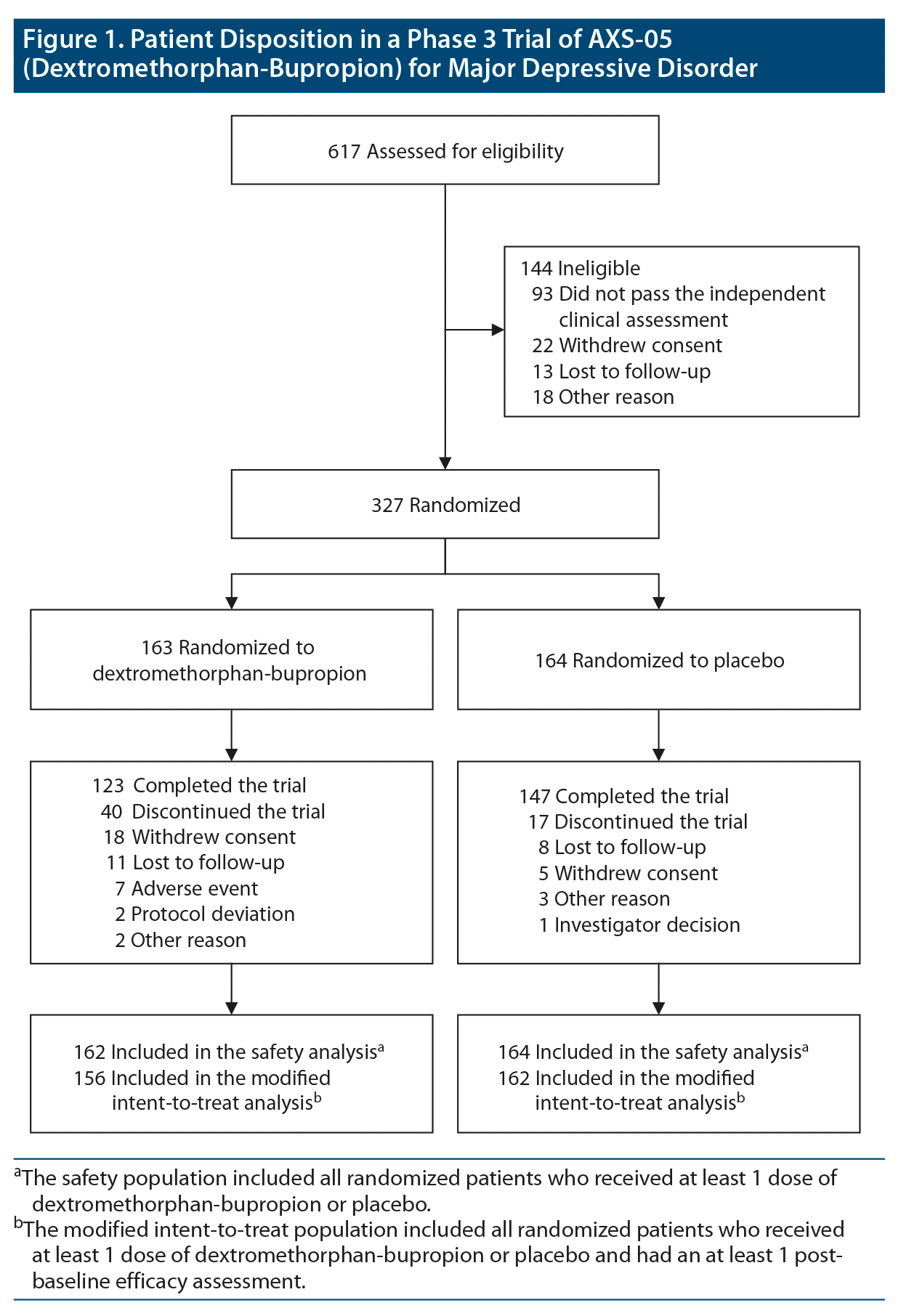
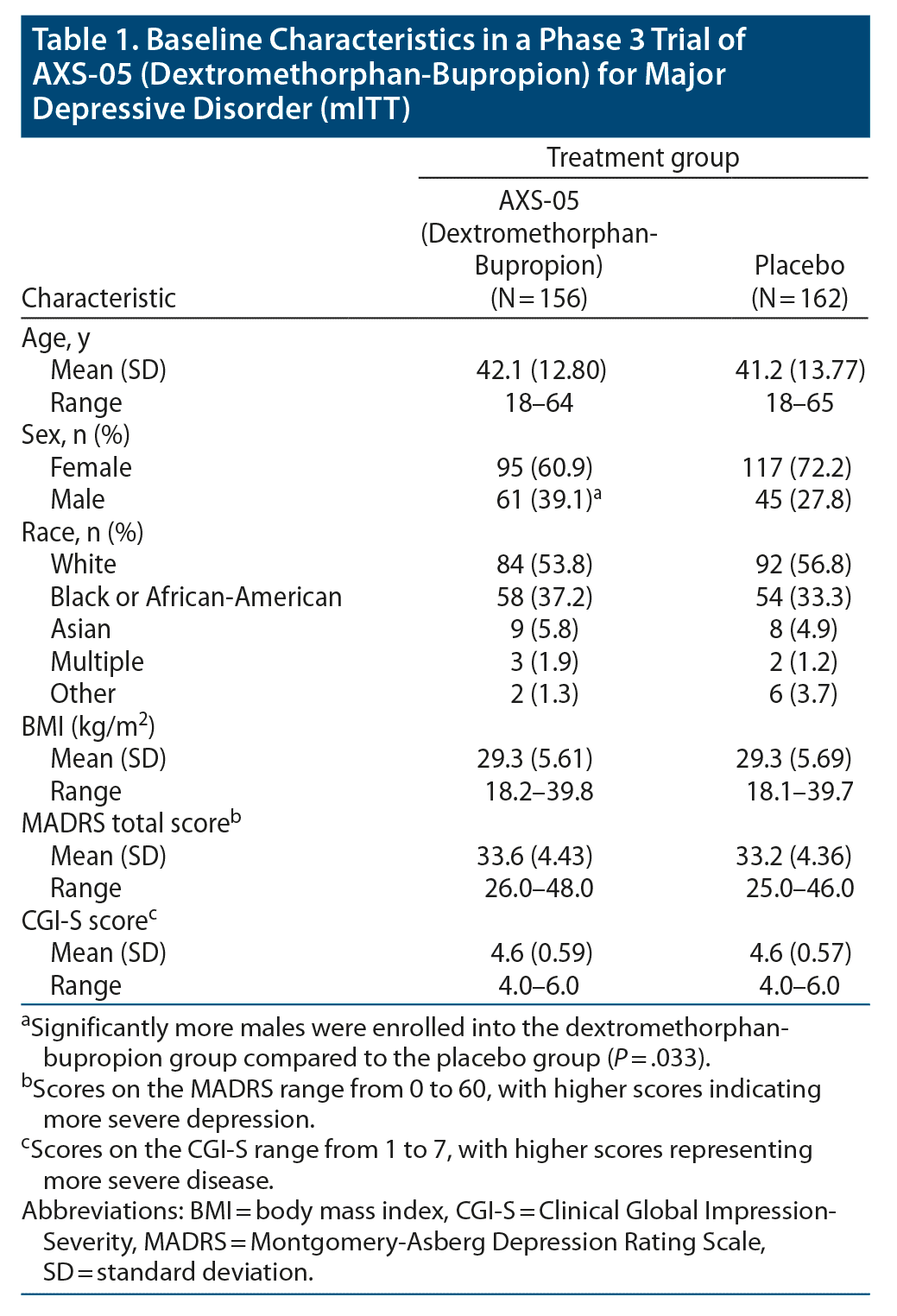
A total of 617 patients were screened, of whom 327 were randomly assigned in a 1:1 ratio to receive dextromethorphan-bupropion (163 patients) or placebo (164 patients) (Figure 1). The modified intent-to-treat population consisted of 156 patients in the dextromethorphan-bupropion group and 162 patients in the placebo group. The demographics and clinical characteristics at baseline were generally similar for the 2 trial groups (Table 1). The dextromethorphan-bupropion group included more men than the placebo group (39.1% vs 27.8%; P = .033). At baseline, mean MADRS total scores were 33.6 in the dextromethorphan-bupropion group and 33.2 in the placebo group, and mean CGI-S scores were 4.6 in both groups. The number of patients who completed the trial was 123 in the dextromethorphan-bupropion group and 147 in the placebo group.
Efficacy
Dextromethorphan-bupropion significantly reduced MADRS total scores compared to placebo at all time points assessed (Figure 2). The least-squares mean change from baseline to week 6 in MADRS total score was −15.9 points in the dextromethorphan-bupropion group and −12.0 points in the placebo group (least-squares mean difference, −3.87; 95% confidence interval [CI], −1.39 to −6.36; P = .002). Results of the sensitivity analyses of the change in MADRS total score from baseline to week 6 were also statistically significant, favoring dextromethorphan-bupropion with a similar magnitude of treatment difference as the primary analysis. At week 1, the first time point, the least-squares mean change from baseline in MADRS total score was −7.20 points in the dextromethorphan-bupropion group and −4.97 points in the placebo group (least-squares mean difference, −2.23; 95% CI, −0.60 to −3.86; P = .007). At week 2, the least-squares mean change from baseline in MADRS total score was −11.09 points in the dextromethorphan-bupropion group and −7.66 points in the placebo group (least-squares mean difference, −3.44; 95% CI, −1.40 to −5.47; P < .001).
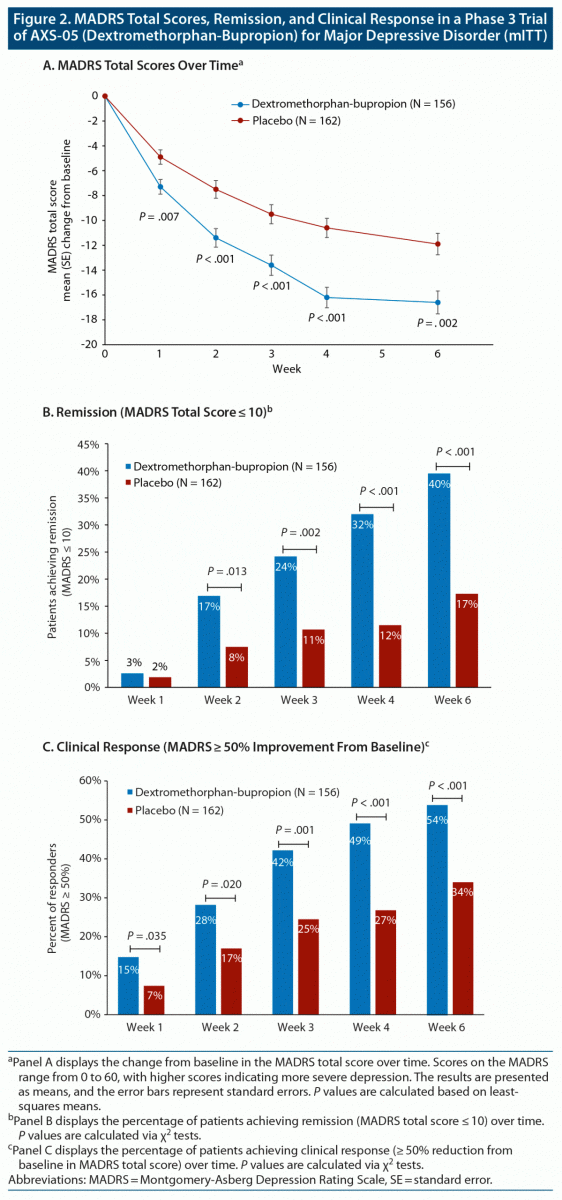
Remission, defined as a MADRS total score of ≤ 10, was achieved by a significantly greater percentage of patients in the dextromethorphan-bupropion group than in the placebo group at week 2 (16.9% and 7.5%, respectively; treatment difference, 9.4%; 95% CI, 1.9%–16.8%; P = .013) and at every time point thereafter. At week 6, the percentage of patients achieving remission was 39.5% in the dextromethorphan-bupropion group and 17.3% in the placebo group (treatment difference, 22.2%; 95% CI, 11.7%–32.7%; P < .001). Clinical response, defined as a ≥ 50% reduction in MADRS total score, was achieved by a significantly greater percentage of patients in the dextromethorphan-bupropion group than in the placebo group at all time points. At week 6, the percentage of patients achieving clinical response was 54.0% in the dextromethorphan-bupropion group and 34.0% in the placebo group (treatment difference, 20.0%; 95% CI, 8.4%–31.6%; P < .001) (Table 2).
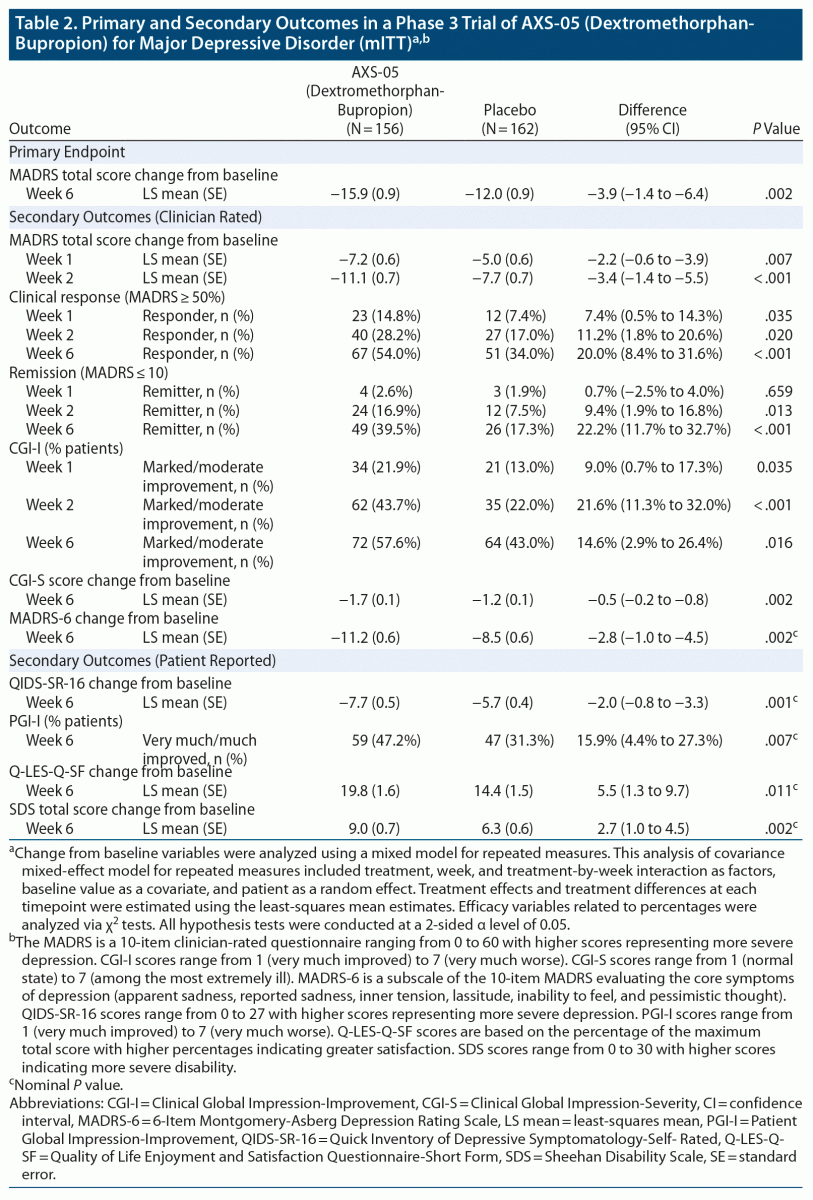
Marked or moderate improvement on the CGI-I was achieved by a significantly higher percentage of patients in the dextromethorphan-bupropion group than in the placebo group at all time points assessed (Table 2). The percentage of patients achieving marked or moderate improvement on the CGI-I at week 6 was 57.6% in the dextromethorphan-bupropion group and 43.0% in the placebo group (treatment difference, 14.6%; 95% CI, 2.9%–26.4%; P = .016). Dextromethorphan-bupropion significantly reduced CGI-S scores compared to placebo at all time points assessed. The least-squares mean change from baseline to week 6 in CGI-S score was −1.69 points in the dextromethorphan-bupropion group and −1.29 points in the placebo group (least-squares mean difference, −0.48; 95% CI, −0.48 to −0.79; P = .002).
At all time points, least-squares mean improvements from baseline on the QIDS-SR-16, the MADRS-6 subscale, and the Q-LES-Q-SF were statistically significantly greater, and the percentage of patients much or very much improved on the PGI-I statistically significantly higher, in the dextromethorphan-bupropion group than in the placebo group. Least-squares mean improvement from baseline on the SDS was statistically significantly greater in the dextromethorphan-bupropion group than in the placebo group at week 2 and at every time point thereafter (Table 2). P values for these endpoints are nominal because of the lack of adjustment for multiplicity for these outcomes.
Safety
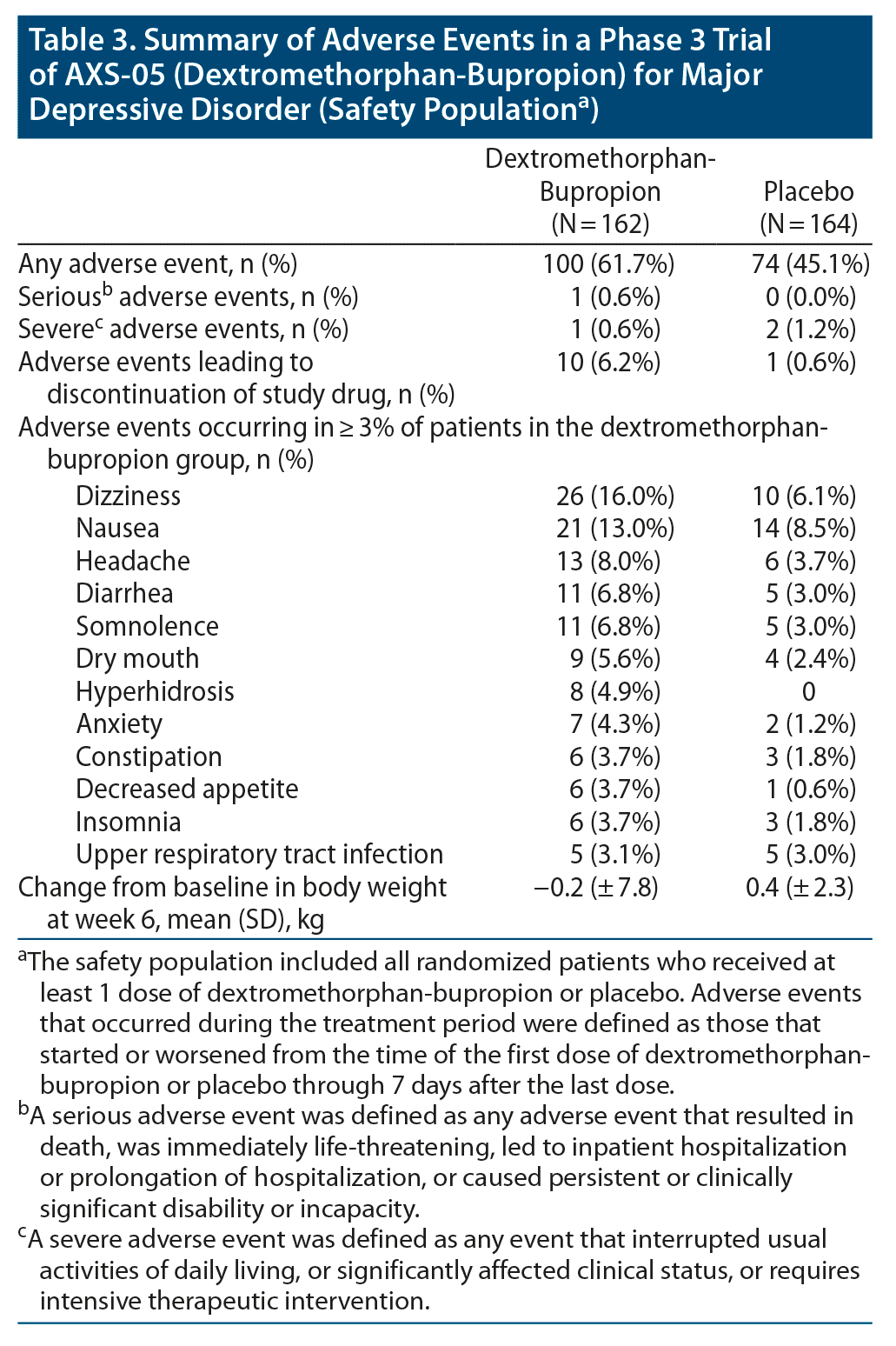
The percentage of patients in whom adverse events occurred during the treatment period was 61.7% in the dextromethorphan-bupropion group and 45.1% in the placebo group. The most common adverse events in the dextromethorphan-bupropion group were dizziness, nausea, headache, somnolence, and dry mouth. There was 1 serious adverse event in the trial, a case of pancreatitis occurring in the dextromethorphan-bupropion group that was determined not related to study medication by the investigator. One adverse event rated severe (migraine) was observed in the dextromethorphan-bupropion group, while 2 events rated severe (back pain, finger fracture repair) were reported in the placebo group. Adverse events resulting in discontinuation of study medication occurred in 6.2% of patients in the dextromethorphan-bupropion group and 0.6% of patients in the placebo group. Dextromethorphan-bupropion was not associated with psychotomimetic effects, weight gain, or increased sexual dysfunction (Table 3). There were no suicide-related adverse events or suicidal behaviors on the C-SSRS in either treatment group. One patient in each treatment group reported suicidal ideation without intent on the C-SSRS at week 6. There were no signals of withdrawal after discontinuation of dextromethorphan-bupropion.
DISCUSSION
In this randomized, placebo-controlled trial, dextromethorphan-bupropion (AXS-05) demonstrated statistically significant antidepressant efficacy on the primary endpoint and the majority of secondary endpoints across multiple symptom-specific and global measures. Treatment with dextromethorphan-bupropion resulted in statistically significantly greater reductions in MADRS total score than placebo starting at week 1 and continuing at every time point thereafter. The medication-placebo difference for dextromethorphan-bupropion on the change in MADRS was substantial at all time points, being approximately 2–3 points at weeks 1 and 2 and increasing to approximately 4–5 points at weeks 4 and 6. This treatment effect compares favorably to the approximately 2.5-point mean difference from placebo seen at 6 to 8 weeks in antidepressant studies in the FDA database.27–29
Symptom remission is considered a desired goal in depression treatment because it is associated with better daily functioning and better long-term prognosis.30 Dextromethorphan-bupropion treatment resulted in early and substantial achievement of remission on the MADRS (total score ≤ 10), with statistically significant separation from placebo demonstrated at week 2 and at every subsequent time point. At week 6, the percentage of patients achieving remission was 39.5% in the dextromethorphan-bupropion group compared to 17.3% in the placebo group (P = .004). Clinical response on the MADRS (≥ 50% reduction from baseline) was also early and substantial with statistical significance versus placebo observed at week 1 and at every subsequent time point. At week 6, the percentage of patients achieving clinical response was 54.0% in the dextromethorphan-bupropion group compared to 34.0% in the placebo group (P < .001).
The early efficacy of dextromethorphan-bupropion was further evidenced by statistically significant benefits versus placebo at week 1 and at all subsequent time points on numerous other clinically relevant measures including MADRS-6, CGI-S, CGI-I, PGI-I, QIDS-SR-16, and Q-LES-Q-SF. Improvement in functional disability was also observed early with statistical significance on the SDS achieved at week 2 and at every time point thereafter.
Dextromethorphan-bupropion was safe and well tolerated in this trial, with low rates of discontinuations due to adverse events. The magnitude of the differences in the rates of adverse events and discontinuations due to adverse events between the two treatment groups in the trial likely reflects the much lower-than-expected rate of these events in the placebo group. For example, the 61.7% overall rate of adverse events in the dextromethorphan-bupropion group is in line with the average of 76.4% reported for antidepressant arms in a large meta-analysis of placebo-controlled depression trials.31 In comparison, the 45.1% rate of adverse events for placebo in this trial is lower than the 63.0% average reported for the placebo arms in the same meta-analysis. Similarly, the 6.2% rate of discontinuations due to adverse events in the dextromethorphan-bupropion group is in line with the average of 7% reported for antidepressant arms in another large meta-analysis of placebo-controlled depression trials.32 In contrast, the 0.6% rate of discontinuations due to adverse events for placebo in our trial is substantially lower than the 4% average reported for the placebo arms in that same meta-analysis. Unlike other NMDA receptor antagonists, dextromethorphan-bupropion was not associated with psychotomimetic effects. This tolerability profile could be related to the significantly faster rate of unblocking of the NMDA receptor channel reported for dextromethorphan as compared to other NMDA antagonists.11 Dextromethorphan-bupropion was not associated with weight gain or increased sexual dysfunction.
Limitations of this trial include exclusion of patients with psychotic or other psychiatric disorders, alcohol/substance use disorders, clinically significant risk of suicide, or significant medical comorbidities. These exclusions along with prohibition of certain concomitant medications may limit the generalizability of the study findings. In addition, treatment at experienced trial sites by specialized clinicians under a trial protocol with frequent clinical assessments may not reflect general practice. The dropout rate of 24.1% in the dextromethorphan-bupropion group is approximately twice that of placebo. However, this rate is in line with the average of 24.3% reported for antidepressant arms in a large meta-analysis of published placebo-controlled depression trials.32 In contrast, the 10.4% dropout rate for placebo in this trial is substantially lower than the 24.0% average reported for placebo arms in the same meta-analysis. Multiplicity adjustments were only applied to the key secondary efficacy endpoints; however, the nominal P values for most other secondary endpoints were < .05. Finally, the trial duration was limited to 6 weeks. The effect of long-term treatment with dextromethorphan-bupropion has been evaluated in a separate study.
In conclusion, this study demonstrated that treatment with dextromethorphan-bupropion (AXS-05) resulted in clinically meaningful and statistically significant improvements in depressive symptoms compared to placebo starting at week 1 in patients with MDD and was well tolerated. The efficacy of dextromethorphan-bupropion was supported by significant improvements compared to placebo on multiple clinically relevant endpoints across symptom-specific and global measures, demonstrating internal consistency of the study results.
Submitted: December 3, 2021; accepted May 5, 2022.
Published online: May 30, 2022.
Author contributions: Concept and design: Iosifescu, Jones, Tabuteau. Acquisition, analysis, or interpretation of data: Iosifescu, Jones, Tabuteau, O’Gorman, Streicher, Feliz, Fava. Drafting of the manuscript: Jones, Tabuteau. Critical revision of the manuscript for important intellectual content: Iosifescu, Jones, Tabuteau, Fava.
Relevant financial relationships: In the last 5 years, Dr Iosifescu has received consulting honoraria from Alkermes, Allergan, Axsome, Biogen, Centers for Psychiatric Excellence, Jazz, Lundbeck, Otsuka, Precision Neuroscience, Sage, and Sunovion and has received research support (through his academic institutions) from Alkermes, AstraZeneca, Brainsway, Litecure, Neosync, Otsuka, Roche, and Shire. Disclosures for Dr Fava are listed at: https://mghcme.org/app/uploads/2021/07/MF-Disclosures-Lifetime-updated-July-2021.pdf. Drs Jones and Tabuteau and Mss Feliz and Streicher are employees of Axsome Therapeutics. Dr O’Gorman is a former employee of Axsome Therapeutics.
Funding/support: This study was funded by Axsome Therapeutics.
Role of the sponsor: Axsome Therapeutics designed the protocol; oversaw the conduct of the study; analyzed and interpreted the data; authored the manuscript; and had final decision to submit the manuscript for publication.
Previous presentation: The results for this study were presented at the American Society of Clinical Psychopharmacology meeting in May 2020 and at the European College of Neuropsychopharmacology in September 2020.
Acknowledgments: The authors thank the participants and investigators who participated in this study. They also thank members of the Axsome Therapeutics Clinical Operations team, including Alexa Blume, BA, and Karry Calderon, BA, for the planning, management, and execution of the clinical trial. Alexa Blume and Karry Calderon are employees of Axsome Therapeutics.
Clinical Points
- Currently available oral antidepressants work primarily via the monoamine pathway, may be associated with partial or inadequate response, and typically take several weeks to produce clinically meaningful effects.
- In this large randomized controlled trial, AXS-05 (dextromethorphan-bupropion), an orally administered NMDA receptor antagonist and σ1 receptor agonist, rapidly reduced depressive symptoms and induced remission in patients with major depressive disorder. AXS-05 was well tolerated.
References (32)

- Saltiel PF, Silvershein DI. Major depressive disorder: mechanism-based prescribing for personalized medicine. Neuropsychiatr Dis Treat. 2015;11:875–888. PubMed
- World Health Organization. Fact Sheets. Depression. 2018.
- Crown WH, Finkelstein S, Berndt ER, et al. The impact of treatment-resistant depression on health care utilization and costs. J Clin Psychiatry. 2002;63(11):963–971. PubMed CrossRef
- Machado-Vieira R, Henter ID, Zarate CA Jr. New targets for rapid antidepressant action. Prog Neurobiol. 2017;152:21–37. PubMed CrossRef
- Fava M, Kendler KS. Major depressive disorder. Neuron. 2000;28(2):335–341. PubMed CrossRef
- Trivedi MH, Rush AJ, Wisniewski SR, et al; STAR*D Study Team. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163(1):28–40. PubMed CrossRef
- Moriguchi S, Takamiya A, Noda Y, et al. Glutamatergic neurometabolite levels in major depressive disorder: a systematic review and meta-analysis of proton magnetic resonance spectroscopy studies. Mol Psychiatry. 2019;24(7):952–964. PubMed CrossRef
- Feyissa AM, Chandran A, Stockmeier CA, et al. Reduced levels of NR2A and NR2B subunits of NMDA receptor and PSD-95 in the prefrontal cortex in major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33(1):70–75. PubMed CrossRef
- Mathews DC, Henter ID, Zarate CA. Targeting the glutamatergic system to treat major depressive disorder: rationale and progress to date. Drugs. 2012;72(10):1313–1333. PubMed CrossRef
- Fava M, Freeman MP, Flynn M, et al. Double-blind, placebo-controlled, dose-ranging trial of intravenous ketamine as adjunctive therapy in treatment-resistant depression (TRD). Mol Psychiatry. 2020;25(7):1592–1603. PubMed CrossRef
- Parsons CG, Quack G, Bresink I, et al. Comparison of the potency, kinetics and voltage-dependency of a series of uncompetitive NMDA receptor antagonists in vitro with anticonvulsive and motor impairment activity in vivo. Neuropharmacology. 1995;34(10):1239–1258. PubMed CrossRef
- Nguyen L, Robson MJ, Healy JR, et al. Involvement of sigma-1 receptors in the antidepressant-like effects of dextromethorphan. PLoS One. 2014;9(2):e89985. PubMed CrossRef
- Niciu MJ, Ionescu DF, Richards EM, et al. Glutamate and its receptors in the pathophysiology and treatment of major depressive disorder. J Neural Transm (Vienna). 2014;121(8):907–924. PubMed CrossRef
- Cobos EJ, Entrena JM, Nieto FR, et al. Pharmacology and therapeutic potential of sigma(1) receptor ligands. Curr Neuropharmacol. 2008;6(4):344–366. PubMed CrossRef
- Pope LE, Khalil MH, Berg JE, et al. Pharmacokinetics of dextromethorphan after single or multiple dosing in combination with quinidine in extensive and poor metabolizers. J Clin Pharmacol. 2004;44(10):1132–1142. PubMed CrossRef
- International Conference on Harmonisation. ICH harmonized tripartite guideline: Guideline for Good Clinical Practice E6(R2). ICH website. https://www.ich.org/page/efficacy-guidelines. 2016. Cited July 2021.
- World Medical Association. WMA Declaration of Helsinki: ethical principles for medical research involving human subjects. WMA website. https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/. 2006.
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134(4):382–389. PubMed CrossRef
- Guy W. ECDEU Assessment Manual for Psychopharmacology. US Department of Health, Education, and Welfare. Public Health Service Alcohol, Drug Abuse, and Mental Health Administration, National Institute of Mental Health, Psychopharmacology Research Branch, Division of Extramural Research Programs; 1976:218–222.
- Diagnostic and Statistical Manual of Mental Disorders. Fifth Edition. American Psychiatric Association; 2013.
- Wu Y, Levis B, Ioannidis JPA, et al; DEPRESsion Screening Data (DEPRESSD) Collaboration. Probability of major depression classification based on the SCID, CIDI, and MINI diagnostic interviews: a synthesis of three individual participant data meta-analyses. Psychother Psychosom. 2021;90(1):28–40. PubMed CrossRef
- Rush AJ, Trivedi MH, Ibrahim HM, et al. The 16-Item Quick Inventory of Depressive Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biol Psychiatry. 2003;54(5):573–583. PubMed CrossRef
- Sheehan DV, Harnett-Sheehan K, Raj BA. The measurement of disability. Int Clin Psychopharmacol. 1996;11(suppl 3):89–95. PubMed CrossRef
- Endicott J, Nee J, Harrison W, et al. Quality of Life Enjoyment and Satisfaction Questionnaire: a new measure. Psychopharmacol Bull. 1993;29(2):321–326. PubMed
- Thase ME, Bowden CL, Nashat M, et al. Aripiprazole in bipolar depression: a pooled, post-hoc analysis by severity of core depressive symptoms. Int J Psychiatry Clin Pract. 2012;16(2):121–131. PubMed CrossRef
- Bech P. Rating scales in depression: limitations and pitfalls. Dialogues Clin Neurosci. 2006;8(2):207–215. PubMed CrossRef
- Khin NA, Chen YF, Yang Y, et al. Exploratory analyses of efficacy data from major depressive disorder trials submitted to the US Food and Drug Administration in support of new drug applications. J Clin Psychiatry. 2011;72(4):464–472. PubMed CrossRef
- Khan A, Bhat A, Kolts R, et al. Why has the antidepressant-placebo difference in antidepressant clinical trials diminished over the past three decades? CNS Neurosci Ther. 2010;16(4):217–226. PubMed CrossRef
- Keefe RS, Kraemer HC, Epstein RS, et al. Defining a clinically meaningful effect for the design and interpretation of randomized controlled trials. Innov Clin Neurosci. 2013;10(suppl A):4S–19S. PubMed
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905–1917. PubMed CrossRef
- Cheung CP, Thiyagarajah MT, Abraha HY, et al. The association between placebo arm inclusion and adverse event rates in antidepressant randomized controlled trials: an examination of the Nocebo Effect. J Affect Disord. 2021;280(Pt A):140–147. PubMed CrossRef
- Schalkwijk S, Undurraga J, Tondo L, et al. Declining efficacy in controlled trials of antidepressants: effects of placebo dropout. Int J Neuropsychopharmacol. 2014;17(8):1343–1352. PubMed CrossRef
This PDF is free for all visitors!