ABSTRACT
Objective: To evaluate response to esketamine nasal spray plus an oral antidepressant (ESK + AD) at day 28 in patients with major depressive disorder (DSM-5) and treatment-resistant depression (TRD) who did not meet response criteria within the first week of treatment.
Methods: The current study is a pooled post hoc analysis of two phase 3, double-blind, active-controlled studies, conducted between August 2015 and February 2018, comparing ESK + AD with an oral antidepressant plus placebo (AD + PBO). Early treatment response was defined as a ≥ 50% decrease in Montgomery-Åsberg Depression Rating Scale total score at day 2 or days 2 and 8. Response rates at day 28 were determined among those not meeting early response criteria.
Results: 518 patients in the analysis had day 28 observations (ESK + AD, n = 310; AD + PBO, n = 208). A greater percentage of patients treated with ESK + AD versus AD + PBO met response criteria beginning at day 2 (17.3% [55/318] vs 9.4% [19/203]) and at all subsequent timepoints, including day 28 (58.7% [182/310] vs 45.2% [94/208]). In day 2 nonresponders, 54.9% vs 44.3% (ESK + AD vs AD + PBO, respectively) achieved response at day 28 (P < .01). Similarly, among day 2 and 8 nonresponders, 52.1% vs 42.4% achieved response by day 28 (P = .01). In nonresponders at day 2 and at days 2 and 8, the odds ratio for a response at day 28 was 1.61 (95% CI, 1.09–2.40) with ESK + AD versus 1.56 (95% CI, 1.04–2.35) with AD + PBO.
Conclusions: Patients with TRD without a demonstrated response within the first week of treatment may still derive benefit from a full 4-week induction course of esketamine nasal spray.
Trial Registration: ClinicalTrials.gov identifiers NCT02417064 and NCT02418585
J Clin Psychiatry 2021;82(4):20m13800
To cite: Turkoz I, Daly E, Singh J, et al. Treatment response with esketamine nasal spray plus an oral antidepressant in patients with treatment-resistant depression without evidence of early response: a pooled post hoc analysis of the TRANSFORM studies. J Clin Psychiatry. 2021;82(4):20m13800.
To share: https://doi.org/10.4088/JCP.20m13800
© Copyright 2021 Physicians Postgraduate Press, Inc.
aJanssen Research & Development, LLC, Titusville, New Jersey
bJanssen Scientific Affairs, LLC, Titusville, New Jersey
cJanssen Research & Development, LLC, San Diego, California
dNow with Neurocrine Biosciences Inc, San Diego, California
eJanssen Research & Development, LLC, Spring House, Pennsylvania
fNow with Acadia Pharmaceuticals Inc, Princeton, New Jersey
gDepartment of Psychiatry and Behavioral Neurobiology, School of Medicine, University of Alabama at Birmingham, Birmingham, Alabama
hDepartment of Psychiatry, School of Medicine, Yale University, New Haven, Connecticut
iDepartment of Psychiatry, University of California San Francisco, San Francisco, California
*Corresponding author: Ibrahim Turkoz, PhD, Department of Statistics & Decision Sciences, Janssen Research & Development, LLC, 1125 Trenton-Harbourton Road, Titusville, NJ 08560 ([email protected]).
Worldwide, major depressive disorder (MDD) is associated with high rates of morbidity, disability, and excess mortality.1–3 In clinical practice, several oral antidepressants are often tried before an adequate option is found. If at least a moderate improvement in symptoms is reported during the initial 4–8 weeks of an antidepressant trial, treatment is usually extended3; however, it may take 10–12 weeks to achieve a full response.3 Approximately one-third of patients with MDD do not achieve an adequate response to multiple antidepressants, and those not responding to ≥ 2 oral antidepressants (given at an adequate dose and duration) in the current major depressive episode (MDE) are considered to have treatment-resistant depression (TRD).4,5 The cumulative amount of time spent on ineffective oral antidepressants is considerable for some patients, and repeated treatment trials can be challenging for patients, their families, and health care providers.3
With the advent of newer augmentation strategies and rapid-acting antidepressant treatments, expectations for visible results within the first week of treatment have also increased.6 Even as many patients may experience clinically meaningful improvement within this timeframe, the time to onset of improvement can be variable for individual patients, as with all antidepressant therapies.7,8 Although the literature is not entirely consistent, observable improvement has been reported over time with rapid-acting therapies, suggesting that lack of response within the first week of treatment is not necessarily predictive of future nonresponse.7–12
Esketamine nasal spray (ESK), a noncompetitive N-methyl-d-aspartate (NMDA) receptor antagonist classified as a rapid-acting agent,6 is indicated for use in conjunction with an oral antidepressant for the treatment of adults with TRD.13 Results of two phase 3 studies in adults with TRD9,14 showed that although most patients treated with ESK + AD (58.7%) met the standard response criteria (ie, ≥ 50% improvement in Montgomery-Åsberg Depression Rating Scale [MADRS] total score) by the end of the full 4-week induction phase of treatment, only a subset of patients (< 20%) achieved this milestone within the first 2 doses (ie, by the end of the first week) of treatment. Therefore, even though ESK + AD demonstrates rapid-acting antidepressant effects in some patients, treatment for the full 4 weeks may still provide additional benefit in those who do not respond within the first 2 doses. The present post hoc analysis aimed to estimate the likelihood of achieving response with ESK + AD and AD + placebo nasal spray (PBO) at day 28 in patients not fulfilling criteria for response at day 2 or at days 2 and 8.
METHODS
Study Design
This was a post hoc analysis of pooled data from TRANSFORM-19 (NCT02417064) and TRANSFORM-214 (NCT02418585), two phase 3, double-blind, active-controlled, multicenter studies of esketamine plus a newly initiated oral antidepressant compared with a newly initiated oral antidepressant plus placebo (Figure 1). The TRANSFORM-1 study was conducted between September 2015 and February 2018, and the TRANSFORM-2 study was conducted between August 2015 and November 2017. Full trial methods are described elsewhere.9,14 Briefly, the studies consisted of a 4-week screening/prospective observation phase, a 4-week double-blind treatment phase, and a follow-up phase lasting up to 24 weeks.
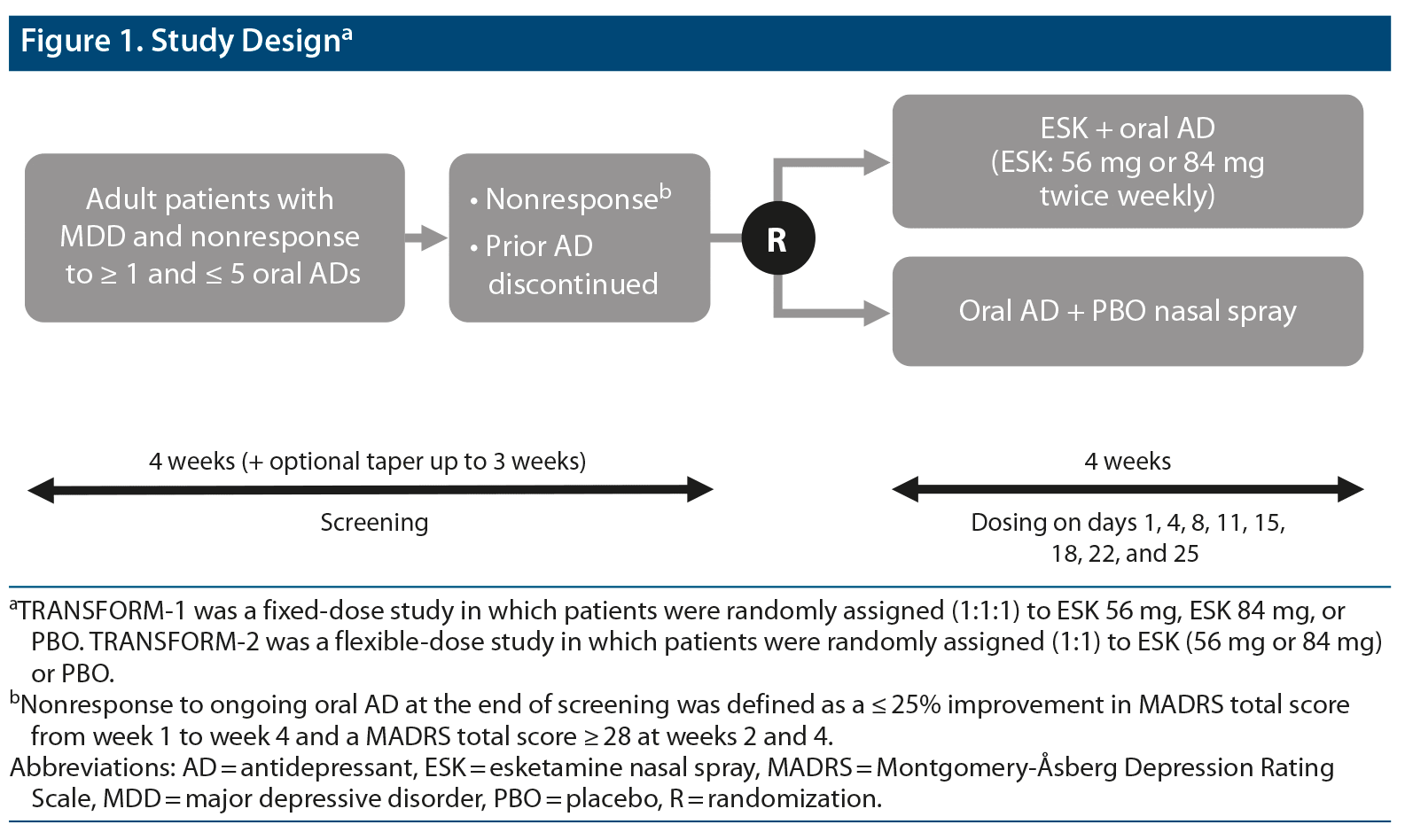
Randomization and Blinding
At the start of the 4-week double-blind treatment phase, a computer-generated randomization schedule was used to randomly assign patients to receive esketamine or placebo. Intranasal study drugs were provided in disposable nasal spray devices with identical appearance and packaging. Each device contained 200-μL solution and delivered 2 sprays of esketamine or placebo. To maintain blinding, the placebo solution had a bittering agent added to simulate the taste of esketamine solution. Patients, investigators, site personnel, and those assessing outcomes and analyzing data were blinded to treatment assignment. As an additional measure to reduce potential impact due to the unique safety profile of esketamine, the primary efficacy measure (MADRS) was conducted by remote, independent raters.
Study Drugs and Administration
In the esketamine arm, patients were randomly assigned to receive twice weekly 1 of 2 fixed doses of esketamine (56 mg or 84 mg) versus placebo (1:1:1) in the TRANSFORM-1 study9 or flexible doses of esketamine (56 mg or 84 mg) versus placebo (1:1) in the TRANSFORM-2 study.14 To increase tolerability, all patients assigned to esketamine treatment started with 56 mg, including those assigned to 84 mg in the fixed-dose study. After day 1, the dose could be titrated to 84 mg per clinical judgment based on efficacy and tolerability. All study patients also received a newly initiated, open-label, oral antidepressant (escitalopram, sertraline, duloxetine, or venlafaxine extended release) taken daily for 4 weeks and titrated to the maximally tolerated labeled dose. Patients self-administered esketamine or placebo twice weekly for 4 weeks at the study site under direct supervision.
Study Population
At study entry, patients were aged 18–64 years with recurrent MDD (per Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition15) or single-episode MDD (≥ 2 years), without psychotic features, confirmed by the Mini-International Neuropsychiatric Interview. Moderate-to-severe depression was confirmed by Inventory of Depressive Symptomatology16 total score ≥ 34 and MADRS17 total score ≥ 28.
At screening, patients were required to have had nonresponse to ≥ 1 but ≤ 5 oral antidepressants, as assessed using the Massachusetts General Hospital Antidepressant Treatment Response Questionnaire. Confirmation of nonresponse to the current ongoing oral antidepressant was required during the following 4-week screening/prospective observation phase. Specifically, at randomization, all patients were required to meet the study definition of TRD: nonresponse to an adequate trial (dose, duration, and adherence) of ≥ 2 antidepressants in the current MDE.
Key exclusion criteria were suicidal ideation with intent to act within the prior 6 months or suicidal behavior within the prior year as assessed by Columbia Suicide Severity Rating Scale (C-SSRS); diagnosis of psychotic disorder, or MDD with psychotic features, bipolar disorder, or related disorders; moderate or severe substance use disorder within the prior 6 months; and positive urine drug test result for specified drugs of abuse at the start of the screening phase or on day 1 of the induction phase (before randomization). Full inclusion/exclusion criteria are described elsewhere.9,14 Institutional review boards or independent ethics committees in Europe, South America, and North America at each study site approved the study protocol and amendments. Studies were conducted in accordance with the ethical principles of the Declaration of Helsinki, Good Clinical Practices, and applicable regulatory requirements. All patients provided written informed consent prior to participation.
The primary efficacy endpoint was change in total MADRS score from baseline to day 28. All MADRS assessments were performed by telephone by independent raters blinded to the protocol details, including study visit, the patient’s clinical status, and adverse events (AEs) occurring during the trial. MADRS was administered with a 24-hour recall period18 at day 2, with a 7-day recall period used at all subsequent assessments.
Statistical Analyses
Analyses were based on the full analysis set, which included all randomized patients who received ≥ 1 dose of study medication and 1 dose of oral antidepressant during the double-blind induction phase. Treatment response was defined as a ≥ 50% improvement in MADRS total score from predose (day 1). For this analysis, 2 definitions of early response were used: ≥ 50% decrease in MADRS total score by day 2 (24 hours after treatment initiation) and ≥ 50% decrease in MADRS total score by day 2 and maintained at day 8 (first week of treatment). Specifically, we sought to determine treatment-related differences in overall response rates and treatment-related differences in the probability of response at day 28 given nonresponse at day 2 or days 2 and 8.
Overall response rates were determined, and treatment group differences were compared using the Cochran-Mantel-Haenszel test controlling for study identification (ID), region, and class of oral antidepressants. Response rates at day 28 were determined among patients without an early response based on these study criteria, and rates were compared between the ESK + AD and AD + PBO groups using various methods to assess the robustness of findings and conclusions. Multiple logistic regression models with key demographic and baseline disease characteristics in addition to study ID and class of oral antidepressants were performed to generate day 28 response probabilities when the study criteria for early response were not met; odds ratios (ORs) comparing treatments and corresponding 95% confidence intervals (CIs) were computed. Multivariate adaptive regression splines (MARS), a nonparametric generalized regression technique combining both splines (nonlinear function) and model selection methods,19,20 was used to estimate day 28 response rates. In addition to logistic regression models, the initial MARS models identified key psychiatric variables associated with response at day 28. Using these covariates, probability of response at day 28 was estimated for both treatment groups, and ORs and 95% CIs were computed. Given that patient-specific responses were measured in a repeated setting, patient-specific conditional probabilities at day 28, if there was no early response, were also computed using repeated-measures generalized linear mixed models. The CIs around predicted probabilities at day 28 were generated based on bootstrapping, which used the estimated variance of random effect for each patient. Predictions from the MARS and repeated measures generalized linear mixed models play a crucial role in assessing the robustness of the findings and conclusions based on empirical scores and logistic regression. These additional analyses provide a diagnostic method to assess the impact, effect, or influence of key assumptions, or variations of different methods of analyses, and to increase confidence in the results.
RESULTS
Patients
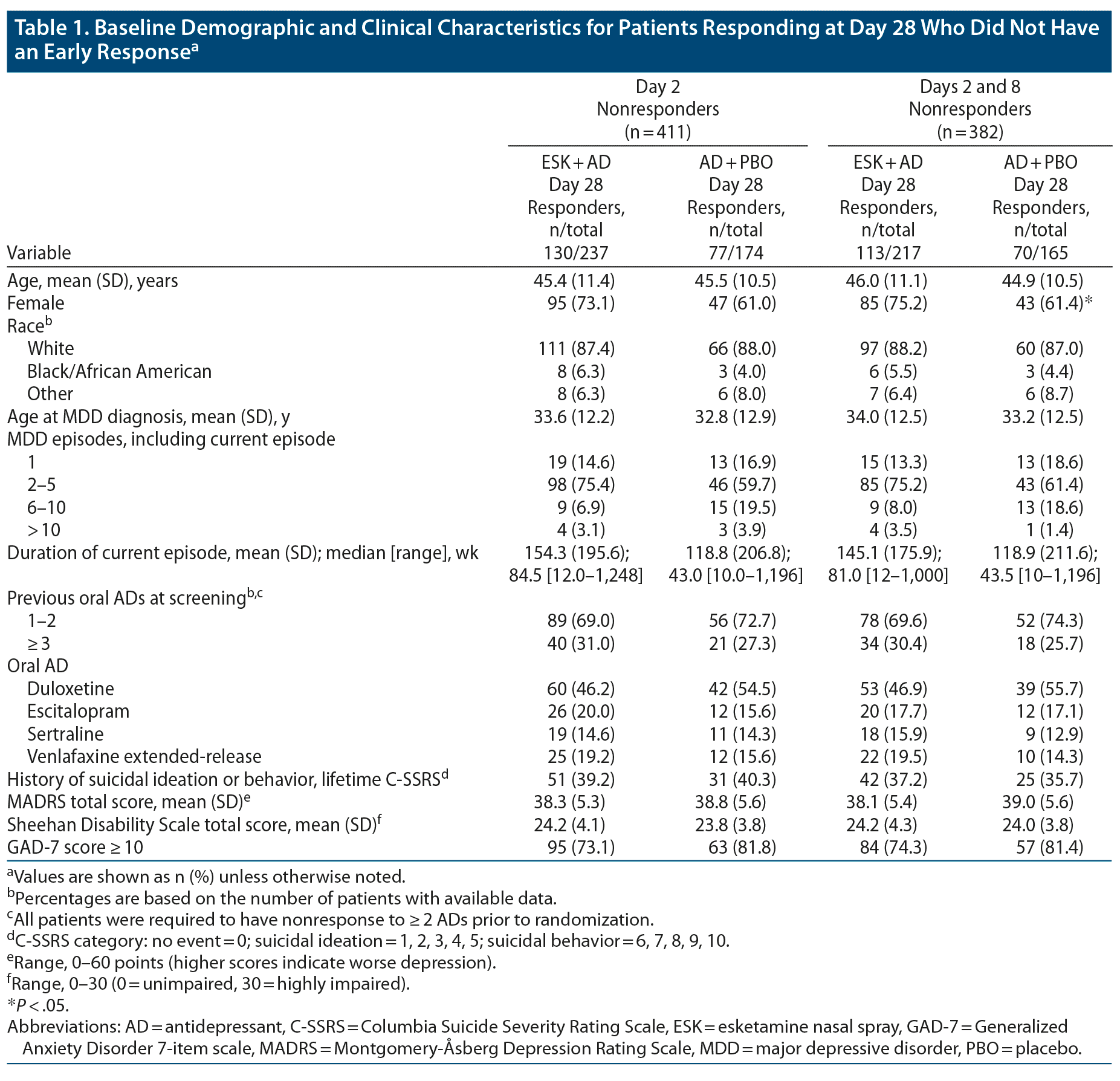
The analysis set included 518 patients with MADRS scores at day 28. No major differences in baseline demographics or psychiatric history were apparent between treatment groups for day 2 nonresponders who responded at day 28. Among day 2 and 8 nonresponders who responded at day 28, there was a significantly higher proportion of female patients in the ESK + AD group compared with the AD + PBO group (Table 1).
Response Rates
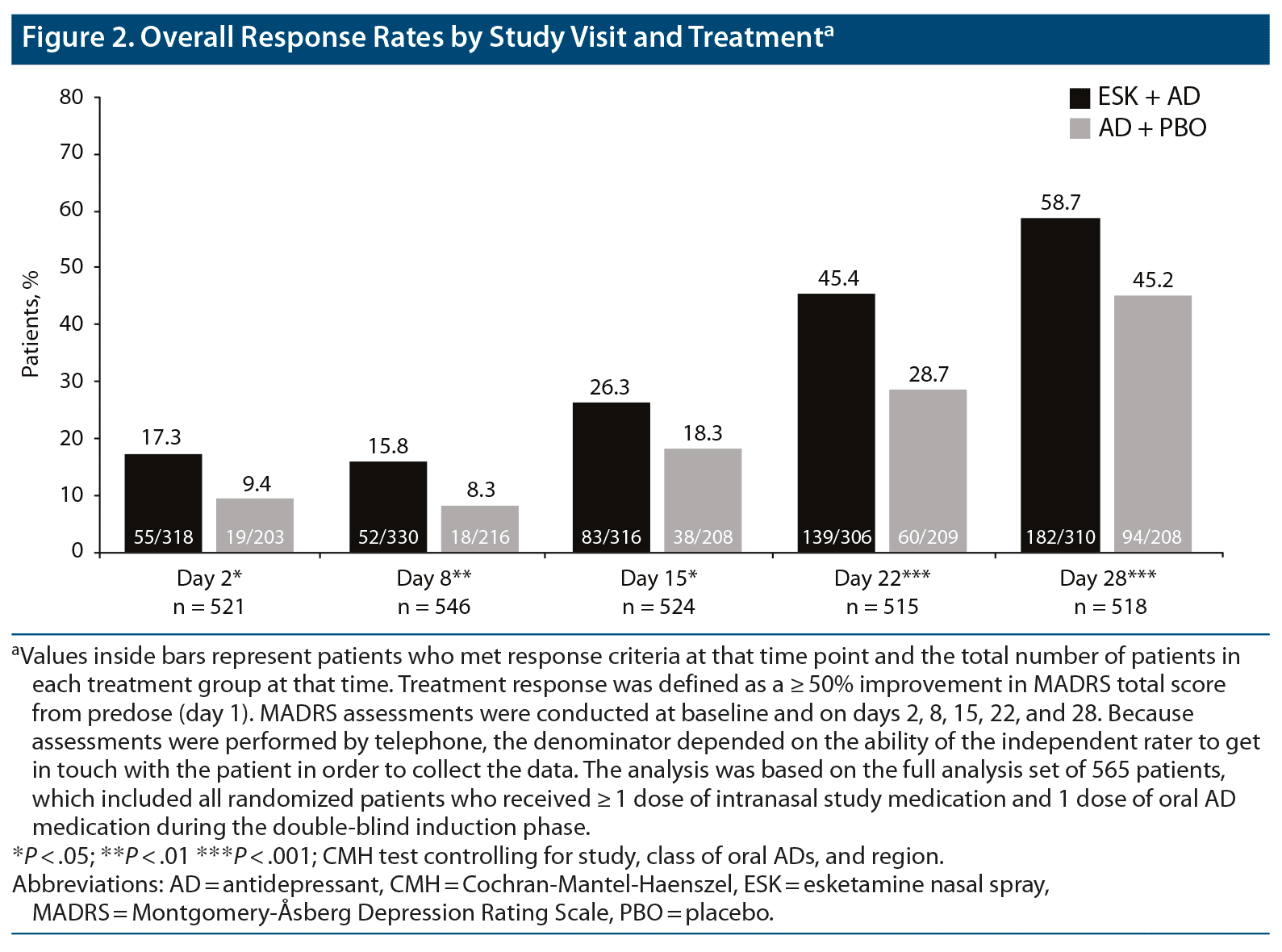
In the full sample, and at each of the 4-week visits, a significantly higher proportion of patients treated with ESK + AD met the criteria for response compared with patients treated with AD + PBO (Figure 2). Differences in response rates between treatment groups differed significantly in favor of ESK + AD at day 2 (17.3% vs 9.4%, ESK + AD vs AD + PBO, P = .01) and at day 8 (15.8% vs 8.3%, ESK + AD vs AD + PBO, P < .01), with 58.7% of ESK + AD patients (vs 45.2% of AD + PBO patients, P < .001) meeting response criteria by day 28.
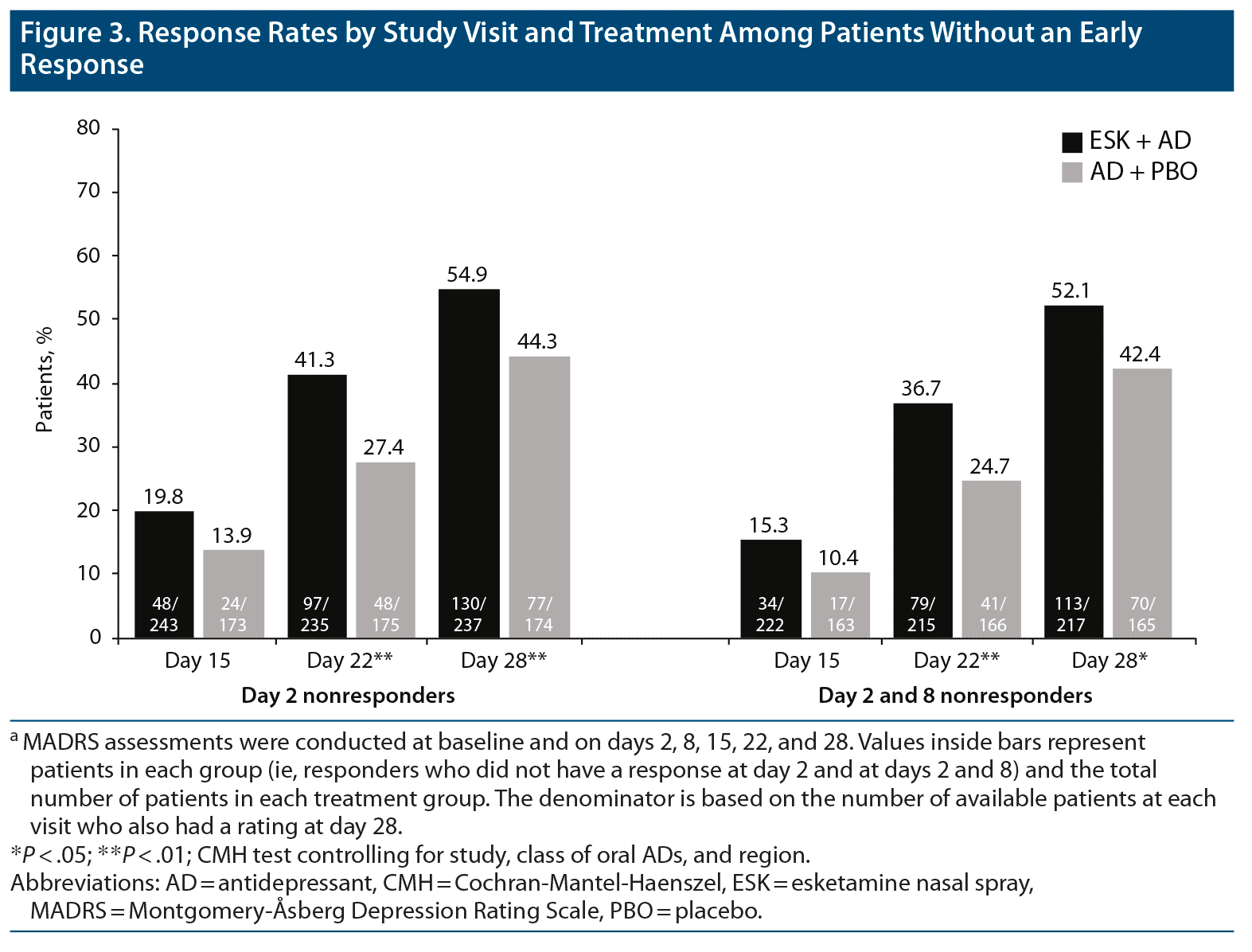
Among both day 2 and day 2 and 8 nonresponders (411 and 382 patients, respectively), the proportion of patients with response at day 28 was significantly higher for ESK + AD than for AD + PBO (Figure 3), similar to findings in the overall population, regardless of early response. In day 2 nonresponders, a response was reported at day 28 in 54.9% and 44.3% of patients treated with ESK + AD and AD + PBO, respectively (P < .01). Similarly, in day 2 and 8 nonresponders, a significantly higher proportion of patients treated with ESK + AD versus AD + PBO reported response at day 28 (52.1% vs 42.4%, respectively; P = .01).
Multiple Logistic Regression Models
Conditional probability computations were carried out using predictors of response at day 28, including baseline MADRS score, lifetime C-SSRS ideation/behavior at screening, and employment status. The predictors of response were initially identified using a stepwise logistic regression model. Among day 2 nonresponders, the OR for response at day 28 with ESK + AD versus AD + PBO was 1.61 (95% CI, 1.09–2.40; probability of response: esketamine, 0.56; placebo, 0.44); for day 2 and 8 nonresponders receiving ESK + AD versus AD + PBO, the OR for response at day 28 was 1.56 (95% CI, 1.04–2.35; probability of response: esketamine, 0.52; placebo, 0.41) (Figure 4A). Therefore, patients treated with ESK + AD had a 61% or 56% increased odds of meeting response criteria at day 28 compared with those treated with AD + PBO who were day 2 nonresponders and day 2 and 8 nonresponders, respectively.

Multivariate Adaptive Regression Spline Models
Relative to logistic regression, MARS models have the benefit of avoiding specific assumptions about the underlying relationship between the dependent and independent variables.20 Logistic regression does not require the dependent and independent variables to be related linearly; however, it requires that independent variables are linearly related to the log odds of response. Factors identified with MARS models as associated with achieving response at day 28 were lifetime C-SSRS ideation/behavior at screening (no event), baseline MADRS total score, and baseline Sheehan Disability Scale score (ie, greater disability associated with lower likelihood of response). After adjusting for these factors in day 2 nonresponders, the OR of response versus nonresponse at day 28 was 1.66 (95% CI, 1.08–2.56), favoring ESK + AD (Figure 4B). Likewise, after adjusting for these factors in day 2 and 8 nonresponders, the OR of response versus nonresponse at day 28 was 1.72 (95% CI, 1.10–2.71), favoring ESK + AD (Figure 4B).
Repeated Measures Generalized Linear Mixed Models
As distinct from the multiple logistic regression and MARS models, the repeated measures generalized linear mixed models allow incorporation of all data points from each visit. Consistent with the previous 2 approaches, results suggest a higher likelihood of achieving response with ESK + AD compared with AD + PBO. In day 2 nonresponders, response was reported at day 28 in 53.3% and 43.3% of patients treated with ESK + AD and AD + PBO, respectively (OR = 1.50; 95% CI, 1.10–2.00). Similarly, in day 2 and 8 nonresponders, a significantly higher proportion of patients treated with ESK + AD versus those treated with AD + PBO reported response at day 28 (49.6% vs 40.1% of patients, respectively; OR = 1.43; 95% CI, 1.04–1.91).
Safety
Treatment-emergent AEs and serious AEs by treatment group are summarized in Supplementary Table 1. Safety and tolerability findings were consistent with primary analyses,9,14 and no new or unexpected safety concerns were noted. Discontinuation rates in day 2 and day 2 and 8 nonresponders over the 4-week study are summarized in Supplementary Table 2.
DISCUSSION
A significantly higher proportion of patients treated with ESK + AD met the criteria for response compared with AD + PBO at day 2 and day 8, with continued higher rates of response throughout the 4-week double-blind treatment phase. In patients without early response at either day 2 or at days 2 and 8, the proportion with response at day 28 was significantly higher in the ESK + AD group than in the AD + PBO group, which has important implications for clinical practice, patient choice, and institutional policies. Based on the present findings and provided that the tolerability profile9,14,21,22 is generally acceptable for individual patients, completion of the full 4-week induction treatment course of esketamine plus an oral antidepressant may be appropriate to optimize treatment effect, even if a full response is not experienced following the first 2 doses of treatment. The present analysis provides an important addition to the limited evidence base, suggesting that later response (defined as a ≥ 50% improvement from baseline in MADRS total score) may be prevalent among patients with TRD receiving treatment with ketamine,7,8 or now, esketamine.
It is notable that the design of the phase 3 esketamine trials in patients with TRD differs from most adjunctive treatment trials conducted in patients with MDD. Esketamine was studied in patients with nonresponse (ie, ≤ 25% improvement since initiation) to ≥ 2 oral antidepressants of adequate dose and duration in the current MDE,9,14 whereas adjunctive therapy is generally used in patients who have experienced a partial response to their current oral antidepressant.3 At the recommendation of the US Food and Drug Administration, to avoid continuing an ineffective oral antidepressant, a new oral antidepressant was initiated at the same time as esketamine or placebo in the phase 3 esketamine trials. This ensured that all patients received clinically optimized and ethical antidepressant treatment. In addition, the frequency of clinical contact (≥ 2 hours twice weekly), which has a demonstrated impact on outcomes,23,24 far exceeded that typically observed in trials of oral antidepressants. In the present analysis, the steep rise in response rates shown in Figure 3, especially in the AD + PBO group, may be related in part to the effects of a new oral antidepressant and/or the increased frequency of clinical contact. Yet, the difference in response rates in initial nonresponding patients appears to increase with time, so that differences between the ESK + AD and AD + PBO groups become larger and significant at weeks 3 and 4.
Finding the optimal antidepressant choice for an individual patient often involves several treatment trials.3 In clinical practice, individualized strategies utilized include dose optimization, switching to a different treatment, combination (simultaneous treatment with 2 antidepressants), augmentation (ie, adjunctive treatment for MDD added to an ongoing oral antidepressant), and use of somatic therapies.25 Nonetheless, as demonstrated in the largest federally funded study of antidepressants,26,27 achieving response or remission in TRD is extremely challenging. Although electroconvulsive therapy, with its response rates of 50%–70%, is recognized as an important treatment for the management of TRD, it carries a risk of cognitive side effects that some patients find unacceptable.28,29 Further research regarding effective management of this population with difficult-to-treat depression is needed.
Early response to ketamine infusion is often cited as a predictor for future response12,30; however, little evidence is available regarding predictors of nonresponse. This analysis contributes to a growing body of evidence that those with early nonresponse may eventually show improvement with continued treatment. In one small, uncontrolled study of repeated-dose ketamine infusion for TRD,7 patients received treatment over 12 days and demonstrated a pattern of continued improvement over the course of the study. Although only 3 patients had achieved response criteria after the first ketamine infusion, 11 of 12 had achieved response by the last of 6 infusions. Another small study8 found similar results, with an average decrease of 2 points on the MADRS with each ketamine infusion and a median of 3 infusions needed to meet response criteria (ie, MADRS score change from baseline ≥ 50%). The present analysis extended prior observations further in that it involved data from 2 active-controlled studies, each with a 4-week double-blind treatment phase, and more than 300 patients who received multiple doses of esketamine, of whom a proportion of patients who did not show an early response at day 2 or days 2 and 8 showed continuous improvement until day 28.
In the current study, multiple analytic approaches provided similar conditional probability of response estimates at day 28 for patients with no response at day 2 or at days 2 and 8. In each case, the likelihood of response with ESK + AD at day 28 was clinically and statistically differentiated from AD + PBO. These results demonstrate that a full 4-week induction treatment course of ESK + AD may lead to greater odds of response compared with AD + PBO in patients with TRD who do not achieve an early full response to treatment as defined in this study. Therefore, if an early response is not observed with esketamine, evidence suggests that continued treatment can still result in a greater likelihood of response than that observed with an antidepressant alone. Ultimately, the decision to continue esketamine treatment is at the discretion of the doctor and patient, and both clinical (eg, safety, tolerability, and efficacy) and practical (eg, time constraints, work demands, and financial means) considerations should be weighed when developing a treatment plan.
Several limitations should be considered when interpreting the results of this study. This post hoc analysis was based on findings from 2 randomized clinical trials with strict inclusion criteria. Consequently, the patient population studied may not fully reflect the diversity of patients with TRD encountered in real-world clinical practice. In these clinical trials, response to treatment was based on stringent, predefined criteria. Therefore, although some patients may have shown evidence of response based on investigator assessment, those not meeting the predefined high threshold for response were not considered “responders” in the present post hoc analysis.
In conclusion, the present results demonstrate that in patients with TRD who do not achieve an early response (defined in this analysis as ≥ 50% improvement in MADRS total score at day 2 or days 2 and 8) to treatment with esketamine nasal spray, a full 4-week induction treatment course of esketamine plus an oral antidepressant can increase the likelihood of response at day 28 compared with an oral antidepressant plus placebo nasal spray. Given the challenges in managing TRD, these findings suggest that patients with TRD may derive benefit from a full 4-week induction treatment course of esketamine nasal spray.
Submitted: November 20, 2020; accepted May 17, 2021.
Published online: July 20, 2021.
Author contributions: Drs Turkoz, Daly, Singh, Lin, Tymofyeyev, Williamson, Salvadore, and Nash had major roles in the conceptualization and oversight of the current post hoc analysis. Drs Daly, Singh, and Salvadore were also directly involved in the design and oversight of the original phase 3 trials in patients with treatment-resistant depression. In addition to contributing to the conceptualization and oversight of the current post hoc analysis, Drs Malacuso and Wilkinson were site investigators who provided direct patient care to clinical trial participants, and Dr Nelson served as chairman for the Independent Safety Monitoring Committee during the phase 3 program. In accordance with International Committee of Medical Journal Editors (ICMJE) guidelines, all authors contributed to the development of the manuscript, approved the final version of the manuscript before submission, and were involved in the decision to submit the manuscript for publication.
Potential conflicts of interest: Drs Turkoz, Daly, Lin, Tymofyeyev, Williamson, and Nash are employees of Janssen and stockholders of Johnson & Johnson, Inc. Dr Salvadore is currently an employee and stockholder of Acadia Pharmaceuticals Inc. At the time that this work was conducted, Dr Salvadore was an employee of Janssen and a stockholder of Johnson & Johnson, Inc. Dr Singh is currently an employee and a stockholder of Neurocrine Biosciences. At the time that this work was conducted, Dr Singh was an employee of Janssen and a stockholder of Johnson & Johnson, Inc. Dr Macaluso has conducted clinical trial research as principal investigator for the following pharmaceutical companies: Acadia, Allergan, Alkermes, Assurex Health/Myriad, Eisai, Lundbeck, Janssen, Neurim, Sage Therapeutics, and Suven. All clinical trial and study contracts were with, and payments made to, the Kansas University Medical Center Research Institute, which is affiliated with his institution, Kansas University School of Medicine–Wichita. In addition, Dr Macaluso is a member of the Janssen speaker bureau. Dr Wilkinson has received contract funding from Janssen, Sage Therapeutics, and Oui Therapeutics for the conduct of clinical trials administered through his institution, Yale University, and has received consulting fees from Janssen, Biohaven, and Oui Therapeutics. Dr Nelson has served as an adviser or consultant to Astellas, Axsome, Biohaven, Janssen, Novartis, Otsuka, and Sunovion.
Funding/support: This research was funded by Janssen Scientific Affairs, LLC.
Role of the sponsor: The sponsor was involved in the design and conduct of the study; collection, analysis, and interpretation of data; preparation, review, and approval of the manuscript; and the decision to submit the manuscript for publication.
Previous presentation: Data reported in this manuscript were previously presented (poster presentation) at the Psych Congress; October 3–6, 2019; San Diego, California.
Acknowledgments: The authors thank the study patients and the investigators for their participation in the studies analyzed. Medical writing and editorial support were provided by Louise Brady, PhD, of ApotheCom (London, UK) and were funded by Janssen Scientific Affairs, LLC. Dr Brady has no conflicts of interest to disclose.
Additional information: Data sharing: The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
Supplementary material: Available at Psychiatrist.com.
Clinical Points
- This post hoc pooled analysis of the TRANSFORM-1 and TRANSFORM-2 studies aimed to estimate the likelihood of achieving response (defined as a ≥ 50% improvement in Montgomery-Åsberg Depression Rating Scale total score) with esketamine nasal spray plus an oral antidepressant after 4 weeks of treatment in adults with treatment-resistant depression who did not have an early response (ie, within the first week).
- Patients who did not achieve an early response to treatment with esketamine nasal spray plus an oral antidepressant had an increased likelihood of achieving response at week 4 compared with those treated with placebo nasal spray plus an oral antidepressant (esketamine nasal spray plus oral antidepressant, 52.1%–54.9%; placebo nasal spray plus oral antidepressant, 42.4%–44.3%)
- Lack of response within the first week of treatment with esketamine plus an oral antidepressant is not necessarily predictive of future nonresponse. These findings suggest that patients with treatment-resistant depression may benefit from the full 4-week induction treatment course of esketamine nasal spray
References (30)

- World Health Organization. Depression. Published January 30, 2020. Accessed June 12, 2020 https://www.who.int/news-room/fact-sheets/detail/depression
- Kessler RC, Bromet EJ. The epidemiology of depression across cultures. Annu Rev Public Health. 2013;34(1):119–138. PubMed CrossRef
- Gelenberg AJ, Freeman MP, Markowitz JC, et al; Work Group on Major Depressive Disorder. Practice Guideline for the Treatment of Patients With Major Depressive Disorder. 3rd ed. Arlington, VA: American Psychiatric Association; 2010.
- Pandarakalam JP. Challenges of treatment-resistant depression. Psychiatr Danub. 2018;30(3):273–284. PubMed CrossRef
- Schwartz J, Murrough JW, Iosifescu DV. Ketamine for treatment-resistant depression: recent developments and clinical applications. Evid Based Ment Health. 2016;19(2):35–38. PubMed CrossRef
- Malhi GS, Morris G, Bell E, et al. A new paradigm for achieving a rapid antidepressant response. Drugs. 2020;80(8):755–764. PubMed CrossRef
- Shiroma PR, Johns B, Kuskowski M, et al. Augmentation of response and remission to serial intravenous subanesthetic ketamine in treatment resistant depression. J Affect Disord. 2014;155:123–129. PubMed CrossRef
- Phillips JL, Norris S, Talbot J, et al. Single, repeated, and maintenance ketamine infusions for treatment-resistant depression: a randomized controlled trial. Am J Psychiatry. 2019;176(5):401–409. PubMed CrossRef
- Fedgchin M, Trivedi M, Daly EJ, et al. Efficacy and safety of fixed-dose esketamine nasal spray combined with a new oral antidepressant in treatment-resistant depression: results of a randomized, double-blind, active-controlled study (TRANSFORM-1). Int J Neuropsychopharmacol. 2019;22(10):616–630. PubMed CrossRef
- El-Khalili N, Joyce M, Atkinson S, et al. Extended-release quetiapine fumarate (quetiapine XR) as adjunctive therapy in major depressive disorder (MDD) in patients with an inadequate response to ongoing antidepressant treatment: a multicentre, randomized, double-blind, placebo-controlled study. Int J Neuropsychopharmacol. 2010;13(7):917–932. PubMed CrossRef
- Kamijima K, Higuchi T, Ishigooka J, et al; ADMIRE Study Group. Aripiprazole augmentation to antidepressant therapy in Japanese patients with major depressive disorder: a randomized, double-blind, placebo-controlled study (ADMIRE study). J Affect Disord. 2013;151(3):899–905. PubMed CrossRef
- Murrough JW, Perez AM, Pillemer S, et al. Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biol Psychiatry. 2013;74(4):250–256. PubMed CrossRef
- SPRAVATO® (esketamine) nasal spray, CIII [prescribing information]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2020.
- Popova V, Daly EJ, Trivedi M, et al. Efficacy and safety of flexibly dosed esketamine nasal spray combined with a newly initiated oral antidepressant in treatment-resistant depression: a randomized double-blind active-controlled study. Am J Psychiatry. 2019;176(6):428–438. PubMed CrossRef
- American Psychiatric Association. Diagnostic and Statistical Manual for Mental Disorders. Fifth Edition. Washington, DC: American Psychiatric Association; 2013.
- Trivedi MH, Rush AJ, Ibrahim HM, et al. The Inventory of Depressive Symptomatology, Clinician Rating (IDS-C) and Self-Report (IDS-SR), and the Quick Inventory of Depressive Symptomatology, Clinician Rating (QIDS-C) and Self-Report (QIDS-SR) in public sector patients with mood disorders: a psychometric evaluation. Psychol Med. 2004;34(1):73–82. PubMed CrossRef
- Williams JB, Kobak KA. Development and reliability of a structured interview guide for the Montgomery-Åsberg Depression Rating Scale (SIGMA). Br J Psychiatry. 2008;192(1):52–58. PubMed CrossRef
- Johnson KM, Devine JM, Ho KF, et al. Evidence to support Montgomery-Åsberg Depression Rating Scale administration every 24 hours to assess rapid onset of treatment response. J Clin Psychiatry. 2016;77(12):1681–1686. PubMed CrossRef
- Hastie T. Additive models, trees, and related methods. In: Hastie T, Tibshirani R, Friedman J, eds. The Elements of Statistical Learning. 2nd ed. Springer Science & Business Media, LLC; 2008:295–336.
- Friedman JH. Multivariate adaptive regression splines. Ann Stat. 1991;19(1):1–67.
- Daly EJ, Trivedi MH, Janik A, et al. Efficacy of esketamine nasal spray plus oral antidepressant treatment for relapse prevention in patients with treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2019;76(9):893–903. PubMed CrossRef
- Wajs E, Aluisio L, Holder R, et al. Esketamine nasal spray plus oral antidepressant in patients with treatment-resistant depression: assessment of long-term safety in a phase 3, open-label study (SUSTAIN-2). J Clin Psychiatry. 2020;81(3):19m12891. PubMed CrossRef
- Cuijpers P, Huibers M, Ebert DD, et al. How much psychotherapy is needed to treat depression? a metaregression analysis. J Affect Disord. 2013;149(1-3):1–13. PubMed CrossRef
- Tiemens B, Kloos M, Spijker J, et al. Lower versus higher frequency of sessions in starting outpatient mental health care and the risk of a chronic course; a naturalistic cohort study. BMC Psychiatry. 2019;19(1):228. PubMed CrossRef
- Al-Harbi KS. Treatment-resistant depression: therapeutic trends, challenges, and future directions. Patient Prefer Adherence. 2012;6:369–388. PubMed CrossRef
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905–1917. PubMed CrossRef
- Trivedi MH, Rush AJ, Wisniewski SR, et al; STAR*D Study Team. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163(1):28–40. PubMed CrossRef
- Wilkinson ST, Agbese E, Leslie DL, et al. Identifying recipients of electroconvulsive therapy: data from privately insured Americans. Psychiatr Serv. 2018;69(5):542–548. PubMed CrossRef
- Greenberg RM, Kellner CH. Electroconvulsive therapy: a selected review. Am J Geriatr Psychiatry. 2005;13(4):268–281. PubMed CrossRef
- Zhou Y, Liu W, Zheng W, et al. Predictors of response to repeated ketamine infusions in depression with suicidal ideation: an ROC curve analysis. J Affect Disord. 2020;264:263–271. PubMed CrossRef
This PDF is free for all visitors!