This Academic Highlights section of The Journal of Clinical Psychiatry presents the highlights of the virtual consensus panel meeting “Dysregulation of Noradrenergic Activity: Its Role in Diagnosing and Treating Major Depressive Disorder, Schizophrenia, Agitation in Alzheimer’s Disease, and Posttraumatic Stress Disorder,” which was held June 5, 2024.
The meeting was chaired by Rakesh Jain, MD, Department of Psychiatry, Texas Tech University School of Medicine-Permian Basin, Midland, Texas. The faculty were Craig Chepke, MD, Excel Psychiatric Associates, Huntersville, North Carolina; Department of Psychiatry, Atrium Health; and Department of Psychiatry, University of North Carolina School of Medicine; Lori L. Davis, MD, Tuscaloosa VA Medical Center, Tuscaloosa, Alabama; Birmingham VA Health Care System, Birmingham, Alabama; and Department of Psychiatry, University of Alabama at Birmingham Heersink School of Medicine, Birmingham; Roger S. McIntyre, MD, Mood Disorder Psychopharmacology Unit, University Health Network, Department of Psychiatry, University of Toronto; Institute of Medical Science, University of Toronto; and Departments of Psychiatry and Pharmacology, University of Toronto, Toronto, Ontario, Canada; Murray A. Raskind, MD, Mental Illness Research, Education, and Clinical Center (MIRECC), VA Puget Sound Health Care System (VA Puget Sound), Seattle, Washington; and Department of Psychiatry and Behavioral Sciences, University of Washington, Seattle.
Financial Disclosures appear at the end of the article
This evidence-based peer-reviewed Academic Highlights was prepared by Healthcare Global Village, Inc. Financial support for preparation and dissemination of this Academic Highlights was provided by H. Lundbeck A/S and Otsuka Product Development and Commercialization. The faculty acknowledges Tony Rodriguez, PharmD, for editorial assistance in developing the manuscript. The opinions expressed herein are those of the faculty and do not necessarily reflect the views of Healthcare Global Village, Inc., the publisher, or the commercial supporters. This article is distributed by H. Lundbeck A/S and Otsuka Product Development and Commercialization for educational purposes only.
J Clin Psychiatry 2024;85(4):plunaro2417ah
Published Online: November 8, 2024.
To Cite: Jain R, Chepke C, Davis LL, et al. Dysregulation of noradrenergic activity: its role in diagnosing and treating major depressive disorder, schizophrenia, agitation in Alzheimer’s disease, and posttraumatic stress disorder. J Clin Psychiatry. 2024;85(4):plunaro2417ah.
To Share: https://doi.org/10.4088/JCP.plunaro2417ah
© 2024 Physicians Postgraduate Press, Inc
When discussing neurotransmitters whose signaling plays an important role in psychiatric illnesses, serotonin and dopamine may be the first that come to mind. Although serotonin and dopamine have significant roles, the impact of norepinephrine signaling is often overlooked. A growing body of evidence suggests that hyperactivity of norepinephrine signaling is an underlying issue in psychiatric disorders; conversely, there is evidence to suggest that deficits in the noradrenergic system are just as significant. Hence, alterations in noradrenergic activity are better characterized as dysregulation rather than a reductive, outdated formulation of “too much” or “too little” activity. Therefore, symptoms such as agitation, irritability, hyperarousal, and insomnia could be treated by targeting the underlying pathophysiology related to noradrenergic dysregulation with targeted treatments. In a recent consensus panel meeting, 5 experts reviewed the available evidence of altered noradrenergic activity and its potential role in some of the most common psychiatric disorders. This Academic Highlights article summarizes their discussion and presents the panel’s conclusions.
Overview of Norepinephrine Pathways and Modulation
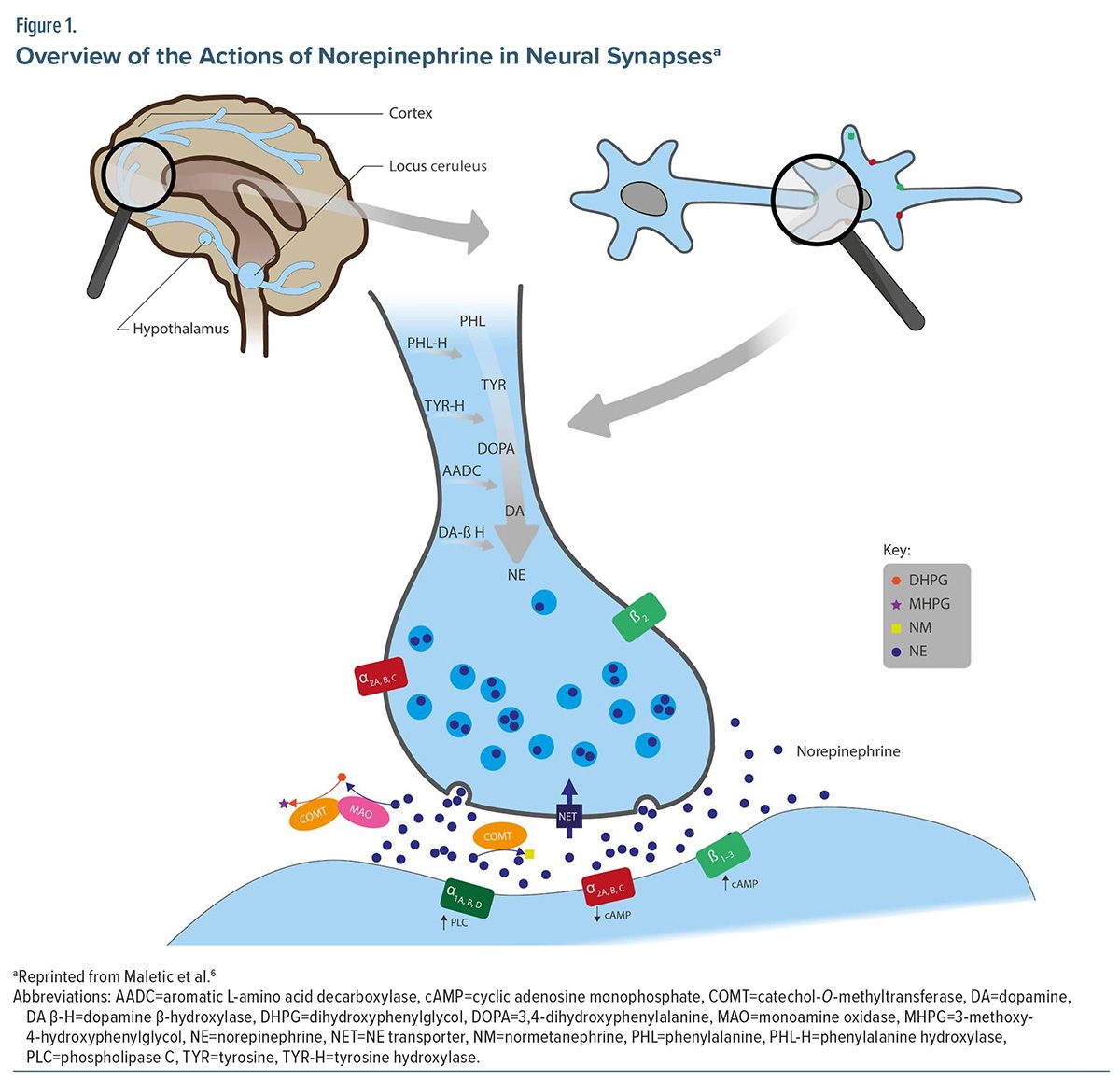
Norepinephrine is a neurotransmitter that plays a significant role in the regulation of mood, level of arousal, cognition, aggression, and awareness via a complex signaling pathway beginning at the locus ceruleus (LC).1,2 The LC is a bilateral nucleus positioned in the upper dorsolateral pontine tegmentum and is responsible for producing the majority of available norepinephrine in the brain. It is widely recognized as the primary noradrenergic nucleus in the central nervous system and serves as a central hub that sends signals to most of the brain. It receives and combines information from several centers responsible for sympathetic, parasympathetic, limbic, and cortical functions. Norepinephrine signaling pathways project widely from the LC to many regions of the brain, including the subcortical and dorsal areas and other neuromodulatory systems (Figure 1).1
Additionally, the LC is interconnected with other systems that modulate neural activity.3 While noradrenergic neurons constitute the most abundant cell population within the LC, GABAergic interneurons are also present and form synapses to provide inhibitory regulation of the activity of noradrenergic neurons. The receptor structure and expression exhibit significant heterogeneity, encompassing adrenergic, GABAergic, serotonergic, glutamatergic, µ-opioid, orexin, nicotinic acetylcholine, and cannabinoid receptors.4
Although the LC does not directly connect to nuclei that are extensively innervated by the dopaminergic system, it can nonetheless impact the distal transmission of dopamine. LC-tyrosine hydroxylase-positive fibers in the cortex, specifically the prefrontal, parietal, and occipital cortices, can release norepinephrine and dopamine, with the release being regulated by alpha-2 adrenoceptors. Activation of the LC also stimulates the release of dopamine in the thalamus and hippocampus, which has a role in stress and cognitive functioning.4 The possibility of additional crosstalk between norepinephrine and other stress responses has been considered. For example, in people with depression, systems like insulin dysregulation can lead to fluctuations in weight and have been associated with worsening of depressive symptoms and suicidality.5
As with all catecholamines, norepinephrine synthesis begins via an amino acid conversion. Phenylalanine is first converted to tyrosine by phenylalanine hydroxylase and then to dihydroxyphenylalanine (DOPA) by tyrosine hydroxylase. DOPA is then converted to dopamine by aromatic L-amino acid decarboxylase and finally to norepinephrine by dopamine beta-hydroxylase. Following synthesis, norepinephrine is stored in presynaptic vesicles before being released into the synaptic cleft via exocytosis. Norepinephrine may be removed from the synaptic space by the norepinephrine transporter, inactivated via the catabolic enzyme catechol-O-methyltransferase, or metabolized by monoamine oxidase. Under normal conditions, this collection of enzymes and receptors plays an important role in regulating the activity of norepinephrine by maintaining an appropriate balance of activity.6
As norepinephrine is released, it can bind to 9 alpha- and beta-noradrenergic receptor subtypes.3 The alpha-1 receptor subtypes include alpha-1a, alpha-1b, and alpha-1d; the alpha-2 receptor subtypes include alpha-2a, alpha-2b, and alpha-2c; and the beta receptor subtypes include beta-1, beta-2, and beta-3. The nomenclature for alpha-1 receptors skips alpha-1c, because receptors initially identified as alpha-1c were later discovered to be identical to alpha-1a receptors.6 The receptor type and location in the synapse, either presynaptically or postsynaptically, determine the signaling effect.7 Receptor subtype density is also variable within brain section, and the receptor’s physiologic effect is variable depending on its location in the brain. For example, alpha-2a receptors are the predominant subtype found in the prefrontal cortex (PFC). However, only small amounts of alpha-2c receptors are found in the PFC.8 Alpha-1 receptor subtypes are expressed in the amygdala, hippocampus, cerebellum, cortex, hypothalamus, and midbrain.9 Consistent with the distribution of alpha-2 receptors, alpha-1a and alpha-1d receptors are predominantly expressed in the PFC; however, the specific location within the PFC has not been determined.8,10 Beta-1 and beta-2 receptors are primarily located in the cerebral cortex and cerebellum and aid in sympathetic nerve activities, while beta-3 receptors are primarily located in brown adipose tissue and urinary detrusor muscle and have less of a role in regulating norepinephrine signaling.11,12
Alpha and beta receptor subtypes located postsynaptically can have either a stimulatory or an inhibitory signaling response, whereas presynaptic receptors exert an inhibitory response by slowing the signaling of norepinephrine into the synaptic space. Postsynaptic alpha-1, beta-1, and beta-2 receptors have stimulatory effects; postsynaptic alpha-1 receptors increase the production and signaling of phospholipase C, whereas postsynaptic beta-1 and beta-2 receptors increase the production and signaling of cyclic adenosine monophosphate (cAMP). These receptors typically interact with Gq proteins, leading to the activation of phospholipase C and phosphatidyl inositol intracellular signaling pathways. This activation then triggers the release of intracellular calcium through inositol 1,4,5-triphosphate (IP3) and the activation of protein kinase C (PKC), ultimately producing an excitatory cellular effect.8,10 Conversely, postsynaptic alpha-2 receptor subtypes decrease the activity of intracellular cAMP by inhibiting specific adenylyl cyclase isoforms, preventing the previously mentioned activation of IP3 and PKC. Presynaptic alpha-2 receptor subtypes and beta-2 receptors decrease the release of norepinephrine into the synaptic cleft via autoreceptor feedback activity.6
Increasing levels of cAMP in the hippocampus allows for improvements in long-term memory, but, conversely, increased cAMP signaling in the PFC has shown impairments in working memory and performance. The upregulation of cAMP production has a vital role in initiating a wide range of cellular responses, such as protein kinase A (PKA), also known as cAMP-dependent protein kinase, cyclic nucleotide-gated channels, or exchange protein activated by cAMP.13 Activated PKA is responsible for adding phosphate groups to various substances found in the cytoplasm and nucleus and ultimately plays an important role in facilitating neuronal plasticity.13 The regulation of cAMP is thought to be impaired in psychiatric disorders like schizophrenia, posttraumatic stress disorder (PTSD), and major depressive disorder (MDD). Decreased cAMP activity in the hippocampus is thought to be directly associated with worse fear memory regulation in people with PTSD, as well as worse anxiety and depressive symptoms in people with schizophrenia and MDD.14–16 Additionally, increases in phospholipase C have been shown to aid in the cellular process of regulating learning and memory in coordination with cAMP.17
Norepinephrine has the highest affinity for alpha-2 receptor subtypes, intermediate binding affinity for alpha-1 receptor subtypes, and the lowest binding affinity for beta receptor subtypes.6,8 Lower levels of norepinephrine will favor inhibitory signaling effects due to the high affinity for alpha-2 receptors. Because alpha-2 receptors on both sides of the synapse will be activated with lower levels of norepinephrine activity, presynaptic alpha-2 receptors create an additional inhibitory effect by decreasing the release of norepinephrine into the synaptic cleft. The rate and timing at which the LC releases norepinephrine, the receptor subtype, and the location to which norepinephrine binds significantly impact an individual’s symptom presentation. Delayed release of norepinephrine to the PFC is directly related to one’s degree of working memory and is potentiated by the alpha-2 receptor.6 When norepinephrine binds to postsynaptic alpha-2 receptors, this allows for improved PFC and hippocampal function, such as working memory and executive functioning.3
It is important to note the impact of receptor downregulation and upregulation on norepinephrine dysregulation. In the absence of norepinephrine, upregulation of both alpha-1 and beta receptors has been observed in hippocampal tissue. Conversely, downregulation of alpha-1 and beta receptors occurs in the setting of an abundance of norepinephrine activity.18 Up- and downregulation of alpha-2 receptors occur linearly relative to the severity and duration of psychosocial stress; upregulation of alpha-2 receptors occurs as the duration and severity of stress increases, and downregulation occurs as stress begins to decrease. This effect is more pronounced for the alpha-2a subtype compared to others.19
Postsynaptic alpha-1 receptors tend to become activated under stress responses when there is increased norepinephrine released into the synaptic space. Their activation is related to increased neuronal activity in the somatosensory cortex.3 Due to having the lowest binding affinity, postsynaptic beta-1 receptor subtypes are activated when there is increased norepinephrine release. When activated, typically under a stress response as well, they support the engagement of sensorimotor cortices, allowing for increased information processing. Under normal conditions, the activation of alpha-2 receptors maintains a homeostatic balance of norepinephrine signaling. During a stress response, activation of alpha-1 receptor subtypes and beta-1 receptors allows for increased sensory and information processing.3 If norepinephrine activity is increased, it could mean overactivation of excitatory alpha-1 and beta-1 receptors, causing a state of hyperarousal, or overactivation of alpha-2 receptors, causing a state of hypoarousal.
While at the receptor level, signaling responses are described as inhibitory or stimulatory, and the resulting behavioral effect depends on the type of receptors inhibited or stimulated. During periods of calm wakefulness, inhibitory signaling of the postsynaptic alpha-2 receptor leads to enhanced rational cognitive functioning at modest concentrations of norepinephrine. During stressful situations, higher concentrations of norepinephrine stimulate alpha-1 receptors, producing the behavioral changes of the fight-or-flight response, such as heightened arousal, wakefulness, increased attention to novel stimuli, and impaired rational working memory. Additionally, higher concentrations of norepinephrine lead to increased peripheral sympathetic nervous system noradrenergic signaling, causing physical symptoms of increased blood pressure, elevated heart rate, and sweating.20
The panel discussed the pharmacology of noradrenergic dysregulation and the importance of norepinephrine as a neurotransmitter target. The panelists offered the analogy of “a tale of two cities”: the problem with norepinephrine in psychiatric disorders is not hyperarousal alone, but, rather, is also hypoarousal and the coexistence of both hyper- and hypoarousal at the same time. There can be rapid fluctuation, or oscillation, of the norepinephrine tone. This may seem paradoxical when compared to nonpsychiatric disorders like diabetes, in which a person cannot be both hyperglycemic and hypoglycemic at the same time.
The panel highlighted that coexisting neurotransmitter dysregulation in opposite directions is present in conditions such as schizophrenia, in which it is well understood that there can be hyperactivity of dopamine signaling in the limbic pathway and hypofunction in the mesocortical pathway.3 Comparing this phenomenon in schizophrenia dopaminergic signaling may help the understanding of how hyper- and hypoarousal due to norepinephrine signaling oscillation can coexist in other conditions.
Panel Consensus Statement #1
Norepinephrine is an important, albeit underappreciated, neurotransmitter with a diverse array of tasks in normal physiological functioning. Dysregulation of the norepinephrine system’s functioning in both the hypo- and hyperfunctionality domains gives rise to numerous manifestations of psychiatric pathology. Norepinephrine has a large number of receptors at its disposal, and today’s clinician should appreciate their diverse roles in modulating norepinephrine functioning.
Hypo- and Hyperactivity Symptoms Related to Norepinephrine Disturbance
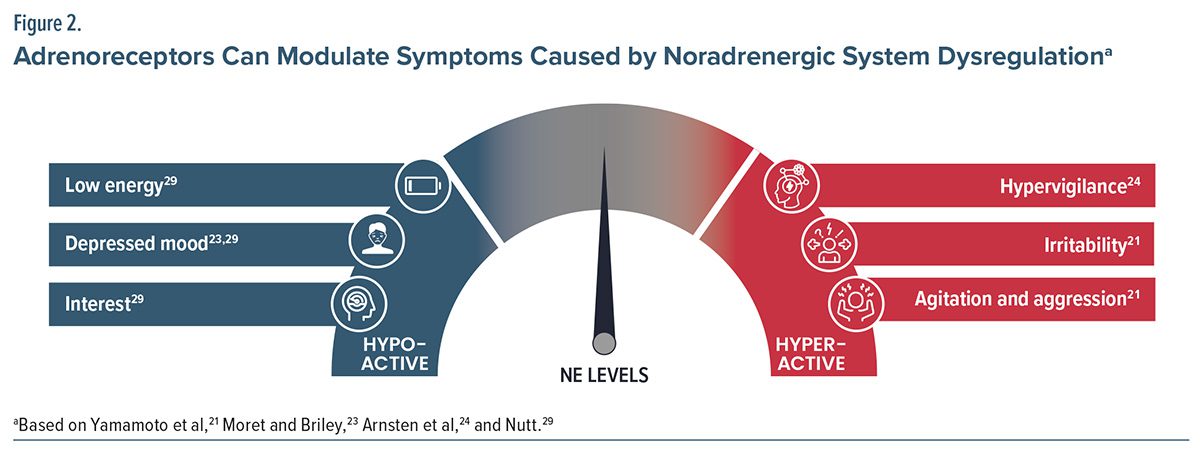
Symptoms from impaired norepinephrine activity stem from both hypo- and hyperactivity. A patient’s level of arousal may be directly correlated to the level of their norepinephrine activity. Table 1 highlights symptoms of overactive and underactive norepinephrine signaling. Overactive norepinephrine signaling may lead to increased sympathetic activity, anhedonia, hyperarousal, insomnia, anxiety, panic, agitation and irritability, fear or paranoia, hypervigilance, distractibility, memory impairment, emotional instability, and aggression. Overactive signaling can also present as the physical symptoms of the fight-or-flight response, including increased blood pressure, heart rate, and sweating. In contrast, hypoactive norepinephrine signaling may lead to decreased sympathetic activity, hypoarousal, poor concentration, hypersomnia, fatigue, lassitude, decreased energy, cognitive slowing, and apathy.21 Little to no activity leads to a rested state with minimal working memory, whereas increased activity leads to a hyperarousal state associated with uncontrollable stress and hypervigilance (Figure 2).22

The panel highlighted the importance of avoiding binary thinking when approaching the assessment of symptoms related to hyperaroused and hypoaroused states. Traditionally, assessments and treatments have focused on either decreasing or increasing norepinephrine activity, which could result in treatment failure because of the potential coexistence of hyperaroused and hypoaroused states in many psychiatric conditions. Because the LC influences norepinephrine release throughout the brain via tonic and phasic firing, norepinephrine activity can be distributed differently in each neuronal body.
A baseline level of arousal is a normal physiologic homeostatic set point. In response to a stressor, one’s level of arousal may adapt and increase. However, if the adaptation is chronic, it can result in a dysregulation of arousal that manifests as psychopathological presentations, as seen in those with attention and anxiety disorders. The tonic, or baseline, firing rate of LC-norepinephrine neurons must be maintained in a specific range of 1 to 3 Hz to support the phasic firing of these neurons.25 Tonic discharge tends to be higher during periods of stress and agitation, moderate during active wakefulness, and lower during drowsiness or sleep. Phasic firing occurs in short bursts at frequencies between 10 to 15 Hz in response to salient stimuli or spontaneously.26 In normal physiology, these two patterns of neuronal activity work together to enable balanced levels of attention. Individuals with hypoarousal experience reduced activity in the LC-norepinephrine system, leading to decreased cortical arousal and impaired attentional performance. Individuals with hyperarousal, such as those with anxiety disorders, experience heightened activity in the LC-norepinephrine system, leading to increased alertness in the brain and reduced ability to focus attention.25
Tonic noradrenergic activity is at its peak level during an active and alert waking state. It gradually drops as the arousal level falls, from quiet waking to a drowsy state and finally to slow-wave sleep. During REM sleep, these neurons are entirely inactive. Phasic activation of the noradrenergic system reaches higher activity levels in response to specific environmental cues. This activation can enhance acute behavioral reactivity to incoming stimuli. Unlike the relationship between tonic noradrenergic activity and arousal, phasic noradrenergic activity is most likely to have widespread acute modulatory effects in response to stress and is strongly activated by various acute stressors. The activation may also be for a brief amount of time or for an extended period of time, depending on the nature of the stressful stimuli. Additionally, during these situations, the activation of norepinephrine due to stress increases the reflex responses of the sensorimotor system.21 While certain responses can be regulated by inducing activity specific to the location and receptor, it is crucial to acknowledge that the noradrenergic system as a whole is activated in the setting of stress. The brain’s noradrenergic system has a unique anatomical organization, responds to various stimuli, and brings about overall changes in brain function during states of heightened alertness. Therefore, if there is an imbalance in norepinephrine signaling, it could potentially contribute to the characteristic symptoms of anxiety disorders and depression. Additionally, changes in brain function in heightened alertness states from pharmacologic treatment could potentially aid in treating these conditions if they restore balanced norepinephrine activity.27
The panel emphasized the relationship between tonic and phasic control of norepinephrine signaling, and that both are negatively impacted in the majority of psychiatric disorders. Additionally, the overlapping symptoms seen in hyperaroused and hypoaroused states are also related to impairments in both tonic and phasic firing. The panel also discussed the importance of clinicians being mindful of the transdiagnostic symptoms that altered norepinephrine activity may cause when approaching the treatment and management of MDD, PTSD, Alzheimer’s disease (AD), and schizophrenia. Additionally, there should be a focus on patients who have not responded to serotonergic interventions, such as those with anxiety, anhedonia, irritability, and ruminative behavior.
Norepinephrine and MDD
Major depressive disorder is known to have a complex etiology involving biological, genetic, environmental, and psychological components and commonly presents with symptoms of irritability, agitation, anxiety, mood, anhedonia, fatigue, somatic symptoms, pain, and rumination of negative thoughts. Previously, MDD was thought to be due to irregularities in neurotransmitters, particularly serotonin, norepinephrine, and dopamine. However, recent hypotheses suggest that it is predominantly linked to more intricate neuroregulatory systems and neuronal circuits, resulting in subsequent disruptions of neurotransmitter systems. The depressive effects of impaired norepinephrine signaling are well known, as serotonin-norepinephrine reuptake inhibitors (SNRIs) have been used in the treatment of MDD for many years.28 SNRIs increase norepinephrine signaling by inhibiting norepinephrine reuptake in the synaptic cleft, allowing for binding to stimulatory receptors.29 However, data from the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial showed that only a minority of patients with MDD will reach symptom remission with first-line therapies.30
The panel emphasized that there is evidence of high and, conversely, low adrenergic signaling that contributes to depressive symptoms and that there may be subgroups of patients with norepinephrine dysregulation that need to be considered when selecting pharmacologic treatment. Additionally, for patients with treatment-resistant depression or patients who failed or had inadequate response to selective serotonin reuptake inhibitor (SSRI) treatment, it may be vital to target modulating norepinephrine activity as part of the treatment protocol. The SNRI venlafaxine has demonstrated significant improvements in anhedonia and amotivation symptoms in those with MDD.31 The US Food and Drug Administration (FDA) and the European Medicines Agency define treatment-resistant depression as the inability to respond to 2 or more antidepressant treatments, even when given at appropriate doses and for a sufficient duration, while also adhering to the prescribed treatment. However, the regulatory bodies acknowledge the imprecision of this concept and its similarity to definitions of “partial response” to antidepressant medication.32 These patients may present an opportunity to evaluate how impaired norepinephrine signaling may influence their symptoms. This is supported by the results of a meta-analysis evaluating the role of norepinephrine dysregulation in MDD and schizophrenia that identified norepinephrine as a possible treatment pathway for patients that require more personalized, targeted therapy, like those with treatment-resistant depression.6
Norepinephrine and PTSD
Since the signs and symptoms of PTSD were codified in DSM-III, it has been hypothesized that distressing PTSD hyperarousal and intrusive symptoms, such as sleep disturbance, irritability, hypervigilance, and trauma content nightmares, reflect persistent and/or inappropriate excessive activation of the noradrenergically mediated components of the “fight or flight” response to environmental threat.33 Over the past 4 decades, multiple neurobiologic studies of the sympathetic nervous system (SNS) and central nervous system (CNS) noradrenergic systems have supported this hypothesis and have provided rationale for antiadrenergic pharmacotherapeutic approaches to PTSD.34
Studies addressing peripheral SNS noradrenergic signaling across multiple parameters are consistent with increased noradrenergic signaling in persons with PTSD compared to controls. These findings include increased alpha-amylase in saliva,35 increased NE in urine,36–38 and increased NE concentrations in plasma at rest and in response to trauma cues.39,40 Because the SNS and CNS systems are co-regulated, these elevated SNS noradrenergic signaling findings in PTSD likely also represent elevated CNS as well as SNS noradrenergic activation. More direct studies of the CNS noradrenergic system also are consistent with increased noradrenergic signaling. Measurements of NE concentrations in cerebrospinal fluid (CSF) in persons with PTSD demonstrate higher CSF NE concentrations at rest41 and trauma reminders.42 Increased pupillary diameter, a correlate of CNS noradrenergic activation, has been demonstrated in PTSD.42 In a recent study using neuromelanin magnetic resonance imaging signal in the LC to provide an integrated measure of LC activity over time, this LC signal was elevated in military veterans with PTSD.43
The repeatedly demonstrated increased noradrenergic signaling in PTSD pathophysiology has provided rationale for an anti-adrenergic approach to pharmacotherapy. This approach has been facilitated by the clinical availability of drugs developed to treat hypertension by reducing noradrenergic signaling. The alpha-2 adrenergic (AR) agonist clonidine and the beta-AR antagonist propranolol appeared helpful for reducing PTSD symptoms in open-label case series.44,45 Unfortunately, parallel-group randomized controlled trials (RCTs) for PTSD of the alpha-2 agonist guanfacine have been negative,46,47 and beta-AR antagonists have not been evaluated in parallel-group RCTs and may actually increase nightmare intensity.
Drugs that antagonize CNS alpha-1 ARs, particularly the brain-penetrant alpha-1 AR antagonist prazosin, as well as the atypical antipsychotics, have been more successful. Prazosin has demonstrated efficacy for PTSD trauma nightmares and hyperarousal symptoms in most, but not all, parallel-group RCTs.48–51 The atypical antipsychotics quetiapine52 and brexpiprazole53 have demonstrated efficacy for reducing PTSD symptoms in RCTs.
Norepinephrine and Agitation in AD
Agitation manifested by irritability, anger outbursts, physical and/or verbal aggression, and pressured motor hyperactivity is a frequent distressing problem for persons with AD and their caregivers54 that is increasingly prevalent as dementia progresses. It is the most common precipitant of placement into long-term care.55 Although agitation in AD is phenomenologically suggestive of excessive CNS noradrenergic signaling, the loss of LC noradrenergic neurons in AD seems inconsistent with this hypothesis. This paradox has been resolved by demonstrations of compensatory upregulation of the CNS noradrenergic system in AD. Concentrations in CSF of NE and its metabolite 3-methoxy-4-hydroxyphenylglycol (MHPG) are elevated in advanced AD.56,57 AD postmortem brain tissue studies demonstrate upregulation of the LC neuronal ability to produce and secrete NE and increased expression of alpha-1 AR in LC projection areas.58–60 Increased behavioral sensitivity to CNS NE in living AD patients has also been demonstrated.61 In that study, increased CNS noradrenergic signaling stimulated by the alpha-2 AR antagonist yohimbine produced equivalently robust increases in CSF NE in AD and normal older comparison participants, but produced agitated behaviors only in the AD group.
The demonstrated increased CNS noradrenergic signaling contribution to agitation in AD provided impetus for trials of postsynaptic AR antagonists to treat agitation in AD. The beta-AR antagonist propranolol effectively reduced agitation compared to placebo in patients with AD, but adverse effects, particularly bradycardia, at doses necessary for reducing agitation over time were problematic.62 A trial of the alpha-1 AR antagonist prazosin for agitation in AD participants in long-term care was more successful,63 with substantial reduction of agitated behaviors in the prazosin vs placebo condition, and was well tolerated with careful dose titration to minimize orthostatic hypotension. Atypical antipsychotics with alpha-1 antagonist activity have been widely prescribed off-label to treat agitation in AD. Secondary analyses of earlier atypical antipsychotic RCTs for “psychosis in AD” as the primary outcome measure have demonstrated efficacy signals for agitation.64 Recent positive trials of the atypical antipsychotic brexpiprazole65 have led to the first FDA-approved drug for the specific indication of agitation in AD.
Norepinephrine and Schizophrenia
Schizophrenia is a highly debilitating mental condition marked by hallucinations and delusions, a lack of drive and emotional responsiveness, and cognitive impairments. Psychosis is the most recognizable symptom of schizophrenia, which is represented by positive symptoms that result in a detachment from reality. These symptoms include hallucinations, delusions, aggression, and disordered thinking. These symptoms are also frequently accompanied by negative symptoms such as diminished emotions, loss of motivation, lack of interest, rumination, insomnia, and disordered conduct. Although there may be differences in how the symptoms appear, the most frequent manifestation in schizophrenia is the presence of paranoid delusions and auditory hallucinations that typically begin in late adolescence or early adulthood, leading to a decline in overall quality of life. Additionally, negative symptoms of schizophrenia are interrelated with traditional manifestations of psychosis and influence one’s decline in quality of life as well.66 Traditionally, the causes of these symptoms have been thought to be due to impairments in the neuromodulation of dopamine, serotonin, and glutamate. However, impairments in norepinephrine activity may also play a significant role in schizophrenia pathophysiology, precipitating cognitive deficits, reducing motivation, and impacting arousal levels. Additionally, cognitive deficits are known to precede schizophrenia onset and predict worse outcomes the more significant the deficits become.3
The Positive and Negative Syndrome Scale (PANSS) is used to measure the severity of 7 positive syndromes, 7 negative syndromes, and 16 general psychopathology symptoms for patients with schizophrenia. Some symptoms assessed by the PANSS may represent symptoms that can be attributed to altered norepinephrine signaling, such as anxiety, agitation, tension due to excessive anxiety or agitation, poor attention or awareness, and alterations in cognitive functioning. Additionally, altered norepinephrine activity may be related to the majority of positive and negative syndromes scored by the PANSS with a positive correlation. Positive symptoms are associated with increased cognitive excitement, grandiosity, hypervigilance, and hostility, which may correlate with increased norepinephrine activity. Conversely, negative symptoms are associated with emotional and social withdrawal, difficulty thinking, and blunted affect, which may be related to decreased norepinephrine activity.
The review of the previously mentioned disorders highlights their similar presenting symptoms and the possible role altered norepinephrine activity may have in each one. To some degree, these symptoms can be graded and possibly attributed to either increased or decreased norepinephrine. It’s important for clinicians to be cognizant of the potential symptoms patients may experience that could be related to impaired norepinephrine activity.
Transdiagnostic Variables
Understanding the network hypothesis can help explain the underlying transdiagnostic neurobiological mechanism that links symptoms of norepinephrine dysregulation across these 4 disease states. Rather than seeking a traditional common root cause for a disease, the network hypothesis emphasizes identifying symptoms that cause other symptoms. Symptoms can be thought of as nodes that interact and connect with other nodes, thus creating a framework in which symptoms like delusion can connect to and create symptoms of paranoia.67,68 The network hypothesis provides a framework to evaluate patterns of connectivity and their dysfunctions between the default mode network (DMN), the central executive network (CEN), and the salience network (SN) which serve as the 3 core neurocognitive networks. The DMN is the primary network during times of cognitive leisure, the CEN is the primary network during times of cognitive exertion, such as times of focus and completing tasks, and the SN serves as a switch between the other two networks. Similar to norepinephrine dysregulation, dysfunctions in SN activity have been correlated to clinical disorders, including anxiety, MDD, insomnia, PTSD, schizophrenia, bipolar disorder, and neurodegenerative illnesses.69,70 Additionally, LC activation of norepinephrine signaling is strongly correlated to network shifting to salience processing and may have a role in the network hypothesis as well. As shown in mouse models, LC activation rapidly interrupts current behavior and significantly increases brain-wide connectivity. This change is significantly associated with norepinephrine release and activation of alpha-1 and beta-1 receptors across the brain.71 Our current understanding of the network hypothesis, along with the LC’s regulation of norepinephrine, further highlights the transdiagnostic nature and shared symptomatology of many psychiatric disorders.
The panel highlighted that trouble initiating sleep or staying asleep has significant potential to both be caused by and further contribute to the dysregulation of norepinephrine signaling and may appear in many psychiatric disorders. Additionally, it is important to know that hypervigilance, hyperarousal, sleep maintenance, REM sleep disruption, and the orexin system are all connected with norepinephrine regulation and that improving sleep quality and sleep hygiene in people with PTSD, MDD, schizophrenia, and AD can aid in balancing norepinephrine activity.
The panel emphasized the importance of clinicians recognizing the impact of symptoms related to impaired norepinephrine activity. Even though the DSM offers a clean division between these disorders, the data and neurobiological studies do not. Patients with psychiatric conditions may be recognized as having comorbidities, but, in reality, these comorbidities may have the same underlying neuropathology. Because clinicians cannot measure levels of norepinephrine in their clinic patients, it is beneficial to assess a patient’s phenotypic information and how it relates to norepinephrine dysregulation. For example, a patient diagnosed with both MDD and PTSD may present with symptoms and meet the diagnostic criteria of each disorder, but one underlying pathology of norepinephrine dysregulation could be responsible for both conditions. Shared symptoms between MDD and PTSD related to norepinephrine dysregulation could include irritability, insomnia, agitation, anxiety, fatigue, and many others related to both hyperaroused and hypoaroused states. The panelists state that the noradrenergic receptors are a fulcrum that can be targeted with receptor-specific medications, but the therapeutic payload is in the network. Utilizing a selective, targeted approach to pharmacologic treatments can improve patient care.
Panel Consensus Statement #2
Norepinephrine dysfunction shows up in a transdiagnostic manner, spanning a variety of seemingly disparate symptoms in a single disorder. For example, norepinephrine may be involved in both the symptoms of fatigue and poor concentration in a patient afflicted with MDD. There is compelling reason to believe norepinephrine disruption is involved in multiple DSM disorders, including MDD, PTSD, schizophrenia, and agitation in AD, among others. It is important to not be monolithic in our conceptualization of the role of various neurotransmitters in these psychiatric pathologies, as norepinephrine is intimately connected to a variety of other neurotransmitters such as glutamate, GABA, serotonin, and dopamine.
The Role of Norepinephrine as a Common Treatment Target
Management of norepinephrine signaling alterations should focus on maintaining a balanced level of norepinephrine activity. Balancing norepinephrine activity can be accomplished by adjusting the extracellular availability of norepinephrine; however, it is possible to overcorrect and cause the reverse effect. Overcorrection of norepinephrine activity in either direction can reverse the symptoms. For example, overcorrection of hyperarousal can lead to hypoarousal and vice versa. Balancing norepinephrine activity can be accomplished by adjusting the availability of norepinephrine in the synaptic cleft, both with and without medications. Medications such as SNRIs are traditionally used to improve the availability of norepinephrine and increase activity. Increasing the amount of norepinephrine available in the synaptic cleft increases norepinephrine’s binding to postsynaptic alpha-2 receptors.6 Some evidence suggests targeting the noradrenergic system can successfully treat AD-related agitation with the use of atypical antidepressants.72 Additionally, since sleep disruptions are a major source of increased norepinephrine activity, improving sleep hygiene can help balance norepinephrine activity.73
As the panel discussed, the focus on modulating norepinephrine activity should be toward the center and requires an understanding of the extracellular availability of norepinephrine, the receptor localization both presynaptically and postsynaptically, and receptor affinity. Because of the varying receptor affinity discussed above, it can be confusing and counterintuitive for clinicians to be told to target the noradrenergic system for both anxious and agitated patients.
Pharmacologic Modulation of Norepinephrine Activity
Pharmacologic management of norepinephrine activity is primarily approached with the use of SNRIs. SNRIs inhibit the presynaptic reuptake of serotonin and norepinephrine from the synaptic cleft.74 Inhibiting the reuptake of norepinephrine allows for increased signaling, which typically increases activity at the stimulatory receptor alpha-2. Based on receptor activity, SNRIs have efficacy in treating symptoms of decreased norepinephrine signaling, such as depressive symptoms, fatigue, and hypoarousal states. Other pharmacologic targets to be considered are beta-blockers and alpha-1 blockers that can cross the blood-brain barrier. These could potentially allow for some blockade of the stimulatory effects of norepinephrine signaling if their receptor affinity is high enough for postsynaptic alpha-1 and beta-1 receptors.75 Additionally, noradrenergic atypical antipsychotics have demonstrated efficacy in decreasing symptoms related to increased norepinephrine activity in AD-related agitation via antagonist activity at alpha-1b and alpha-2c receptors. However, this is specific to broad-spectrum atypical antipsychotics that can affect norepinephrine modulation both presynaptically and postsynaptically.72
The panel discussion highlighted the counterintuitive nature of targeting the noradrenergic system in patients with anxiety, agitation, or fatigue, as modulation of norepinephrine activity without targeted treatments may lead to overcorrection and create opposing symptoms. The focus should be on targeted treatments, identifying patients’ phenotypic symptoms, and the neurobiological evidence of the responsible receptors. However, it may be confusing or counterintuitive to try and target any system in a patient experiencing anxiety or agitation. The panel provides an analogy that only braking or accelerating will never make a car reach its safe location, but, rather, both a functioning brake and an accelerator mechanism are needed to function appropriately. Targeted treatments for conditions with underlying dysregulation of noradrenergic signaling would be able to affect both the “accelerating” and “braking” mechanisms in the neuronal system pharmacologically better to mimic the body’s homeostatic regulation of norepinephrine firing. The most commonly used agents, including norepinephrine reuptake inhibitors, are too nonspecific, preventing norepinephrine reuptake everywhere and flooding the brain with norepinephrine. Conversely, some direct norepinephrine receptor modulators like prazosin, clonidine, and guanfacine, which target only the braking or the accelerating mechanisms, may be too narrow. As such, treatment goals should focus on the ability to modulate the symptoms created both by hypernoradrenergia and hyponoradrenergia safely and appropriately.
Targeted, precision therapeutic options for norepinephrine modulation would be the ideal therapeutic choice if available. Clinicians should approach norepinephrine modulators wisely, as modulation may worsen symptoms if overcorrection occurs or if norepinephrine modulation is too broad pharmacologically. The panel agreed that, if done wisely, the use of norepinephrine modulators is recommended and stated, “Our focus should be on precision therapeutics in our treatment efforts rather than broad spectrum, nonspecific interventions.”
Panel Consensus Statement #3
The norepinephrine system’s dysregulation is squarely at the center of a large number of psychiatric disorders and psychopathological symptoms. For decades, norepinephrine has been in the shadows, but recent research is bringing it center stage in our clinical conceptualization of various psychopathologies. Treatment of various disorders should consider norepinephrine modulation as one of the goals of treatment, while keeping in mind that simplistic conversations of decreasing or increasing norepinephrine levels are now scientifically antiquated. Instead, today’s clinician is called to think astutely and thoughtfully about the optimum modulation of norepinephrine functioning through a wise combination of pre- and post-synaptic receptor actions. The goal of treatment will always remain the minimization of side effects and maximization of clinical outcomes in the short and long run holistic treatment of patients.
Conclusions
As the panel emphasized, the focus on altered norepinephrine signaling involves 3 parts: the extracellular availability of norepinephrine, receptor localization both pre- and postsynaptically, and receptor affinity. Treatments focusing on maintaining a balance of norepinephrine activity are crucial since symptoms can develop at both high and low levels of activity because of the varying receptor affinity and activation at different levels of norepinephrine activity. Lastly, it is important to be aware of the nuanced role of norepinephrine activity in these disease states.
Financial Disclosures
Dr Jain has received consulting fees from Abbvie, Acadia, Adamas, Alfasigma, Alkermes, Almatica, Axsome, Biogen, Boehringer Ingelheim, Corium, Cingulate Therapeutics, Eisai, Evidera, Impel, Janssen, Lilly, Lundbeck, Merck, Neos Therapeutics, Neurocrine Biosciences, Osmotica, Otsuka, Pamlab, Pfizer, Sage Therapeutics, Shire, Sumitomo, Sunovion, Supernus, Takeda, Teva, Transcend Therapeutics, and Viatris; grant/research support from Abbvie (Allergan), Lilly, Lundbeck, Otsuka, Pfizer, Shire, and Takeda; honoraria for speaking/teaching from Abbvie, Alkermes, Almatica, Axsome, Corium, Eisai, Janssen, Lilly, Lundbeck, Merck, Neos Therapeutics, Neurocrine, Otsuka, Pamlab, Pfizer, Shire, Sunovion, Takeda, Teva, Tris Pharmaceuticals, and Viatris; and advisory board fees from Adamas, Alkermes, Corium, Eisai, Janssen, Lilly, Lundbeck, Merck, Neos Therapeutics, Neurocrine Biosciences, Otsuka, Pamlab, Pfizer, Sage Therapeutics, Shire, Sunovion, Supernus, Takeda, Teva, and Usona. Dr Chepke has received consulting fees from Abbvie, Acadia, Alkermes, Axsome, Biogen, Boehringer Ingelheim, Corium, Intra-Cellular, Janssen, Karuna, Lundbeck, MedinCell, Moderna, Neurocrine, Noven, Otsuka, Sage, Sumitomo, and Teva; grant/research support from Acadia, Axsome, Harmony, Neurocrine, and Teva; honoraria for speaking/teaching from Abbvie, Acadia, Alkermes, Axsome, Corium, Intra-Cellular, Janssen, Karuna, Lundbeck, Merck, Neurocrine, Noven, Otsuka, Sage, Sumitomo, and Teva; and advisory board fees from Abbvie, Acadia, Alkermes, Axsome, Biogen, Corium, Idorsia, Intra-Cellular, Janssen, Karuna, Lundbeck, Moderna, Neurocrine, Noven, Otsuka, Sage, Sumitomo, and Teva. Dr Davis has received consulting fees from Otsuka and Boehringer Ingelheim; grant/research support from VA, Alkermes, Social Finance, and Department of Defense; honoraria for speaking/teaching from Clinical Care Options; and advisory board fees from Otsuka and Boehringer Ingelheim. Dr McIntyre has received speaking/consulting fees from Lundbeck, Janssen, Alkermes, Neumora Therapeutics, Boehringer Ingelheim, Sage, Biogen, Mitsubishi Tanabe, Purdue, Pfizer, Otsuka, Takeda, Neurocrine, Sunovion, Bausch Health, Axsome, Novo Nordisk, Kris, Sanofi, Eisai, Intra-Cellular, NewBridge Pharmaceuticals, Viatris, Abbvie, and Atai Life Sciences; has received grant/research support from CIHR/GACD/National Natural Science Foundation of China, and the Milken Institute; and is the CEO of Braxia Scientific Corp. Dr Raskind reports no relevant financial relationships.
References (75)

- Bari BA, Chokshi V, Schmidt K. Locus coeruleus-norepinephrine: Basic functions and insights into Parkinson’s disease. Neural Regen Res. 2020;15(6):1006–1013. PubMed CrossRef
- Tanaka M, Yoshida M, Emoto H, et al. Noradrenaline systems in the hypothalamus, amygdala and locus coeruleus are involved in the provocation of anxiety: Basic studies. Eur J Pharmacol. 2000;405(1–3):397–406. PubMed CrossRef
- Mäki-Marttunen V, Andreassen OA, Espeseth T. The role of norepinephrine in the pathophysiology of schizophrenia. Neurosci Biobehav Rev. 2020;118:298–314. PubMed CrossRef
- Paredes-Rodriguez E, Vegas-Suarez S, Morera-Herreras T, et al. The noradrenergic system in Parkinson’s disease. Front Pharmacol. 2020;11:435. PubMed CrossRef
- McIntyre RS. The co-occurrence of depression and obesity: Implications for clinical practice and the discovery of targeted and precise mechanistically informed therapeutics. J Clin Psychiatry. 2024;85(2). PubMed CrossRef
- Maletic V, Eramo A, Gwin K, et al. The role of norepinephrine and its α-adrenergic receptors in the pathophysiology and treatment of major depressive disorder and schizophrenia: A systematic review. Front Psychiatry. 2017;8:42. PubMed CrossRef
- Suttkus S, Schumann A, de la Cruz F, et al. Working memory in schizophrenia: The role of the locus coeruleus and its relation to functional brain networks. Brain Behav. 2021;11(5). PubMed CrossRef
- Ramos BP, Arnsten AF. Adrenergic pharmacology and cognition: Focus on the prefrontal cortex. Pharmacol Ther. 2007;113(3):523–536. PubMed CrossRef
- Perez DM. Α(1)-adrenergic receptors in neurotransmission, synaptic plasticity, and cognition. Front Pharmacol. 2020;11:581098. PubMed CrossRef
- Birnbaum SG, Yuan PX, Wang M, et al. Protein kinase C overactivity impairs prefrontal cortical regulation of working memory. Science. 2004;306(5697):882–884. PubMed CrossRef
- Kuhar M, Couceyro P, Lambert P. Basic neurochemistry: Molecular, cellular and medical aspects: Α- and β-adrenergic receptors. Updated 1999. Accessed June 25. PubMed
- Oshima N, Onimaru H, Yamamoto K, et al. Expression and functions of β1- and β2-adrenergic receptors on the bulbospinal neurons in the rostral ventrolateral medulla. Hypertension Res. 2014;37(11):976–983. PubMed CrossRef
- Argyrousi EK, Heckman PRA, Prickaerts J. Role of cyclic nucleotides and their downstream signaling cascades in memory function: Being at the right time at the right spot. Neurosci Biobehav Rev. 2020;113:12–38. PubMed CrossRef
- Fujita M, Richards EM, Niciu MJ, et al. cAMP signaling in brain is decreased in unmedicated depressed patients and increased by treatment with a selective serotonin reuptake inhibitor. Mol Psychiatry. 2017;22(5):754–759. PubMed CrossRef
- Hori H, Fukushima H, Nagayoshi T, et al. Fear memory regulation by the cAMP signaling pathway as an index of reexperiencing symptoms in posttraumatic stress disorder. Molecular Psychiatry. 2024. PubMed CrossRef
- Wang M, Ramos BP, Paspalas CD, et al. Α2a-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129(2):397–410. PubMed CrossRef
- Fulton D, Condro MC, Pearce K, et al. The potential role of postsynaptic phospholipase C activity in synaptic facilitation and behavioral sensitization in aplysia. J Neurophysiol. 2008;100(1):108–116. PubMed CrossRef
- Mynlieff M, Curella P, Zahniser NR, et al. Regulation of adrenergic receptors in intraocular hippocampal transplants: Role of noradrenergic innervation. Synapse. 1990;6(2):113–120. PubMed CrossRef
- Flügge G, Van Kampen M, Meyer H, et al. Α2a and α2c-adrenoceptor regulation in the brain: Α2a changes persist after chronic stress. European Journal of Neuroscience. 2003;17(5):917–928. PubMed CrossRef
- Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009;10(6):410–422. PubMed CrossRef
- Yamamoto K, Shinba T, Yoshii M. Psychiatric symptoms of noradrenergic dysfunction: A pathophysiological view. Psychiatry Clin Neurosci. 2014;68(1):1–20. PubMed CrossRef
- Yücel M, Oldenhof E, Ahmed SH, et al. A transdiagnostic dimensional approach towards a neuropsychological assessment for addiction: an international Delphi consensus study. Addiction. 2019 Jun;114(6):1095–1109. PubMed
- Moret C, Briley M. The importance of norepinephrine in depression. Neuropsychiatr Dis Treat. 2011;7(Suppl 1):9–13.
- Arnsten AF, Raskind MA, Taylor FB, Connor DF. The effects of stress exposure on prefrontal cortex: translating basic research into successful treatments for post-traumatic stress disorder. Neurobiol Stress. 2015;1:89–99.
- Howells FM, Stein DJ, Russell VA. Synergistic tonic and phasic activity of the locus coeruleus norepinephrine (LC-NE) arousal system is required for optimal attentional performance. Metabol Brain Dis. 2012;27(3):267–274. PubMed CrossRef
- Janitzky K. Impaired phasic discharge of locus coeruleus neurons based on persistent high tonic discharge—a new hypothesis with potential implications for neurodegenerative diseases. Front Neurol. 2020;11:371. PubMed CrossRef
- Morilak DA, Frazer A. Antidepressants and brain monoaminergic systems: A dimensional approach to understanding their behavioural effects in depression and anxiety disorders. Int J Neuropsychopharmacol. 2004;7(2):193–218. PubMed CrossRef
- Kennedy SH. Core symptoms of major depressive disorder: Relevance to diagnosis and treatment. Dialogues Clin Neurosci. 2008;10(3):271–277. PubMed CrossRef
- Nutt DJ. Relationship of neurotransmitters to the symptoms of major depressive disorder. J Clin Psychiatry. 2008;69 Suppl E1:4–7.
- Sinyor M, Schaffer A, Levitt A. The Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial: a review. Can J Psychiatry. 2010 Mar;55(3):126–35. PubMed
- McIntyre RS, Agid O, Biesheuvel E, et al. Effect of venlafaxine on anhedonia and amotivation in patients with major depressive disorder. CNS Spectr. 2024 Jun;29(3):206–214. PubMed
- McIntyre RS, Alsuwaidan M, Baune BT, et al. Treatment-resistant depression: Definition, prevalence, detection, management, and investigational interventions. World Psychiatry. 2023;22(3):394–412. PubMed CrossRef
- Southwick SM, Krystal JH, Morgan CA, et al. Abnormal noradrenergic function in posttraumatic stress disorder. Arch Gen Psychiatry. 1993 Apr;50(4):266–74. PubMed
- Hendrickson RC, Raskind MA. Noradrenergic dysregulation in the pathophysiology of PTSD. Exp Neurol. 2016 Oct;284(Pt B):181–195. PubMed
- Keeshin BR, Strawn JR, Out D, et al. Elevated salivary alpha amylase in adolescent sexual abuse survivors with posttraumatic stress disorder symptoms. J Child Adolesc Psychopharmacol. 2015 May;25(4):344–50. PubMed CrossRef
- Kosten TR, Rounsaville BJ, Babor TF, et al. Substance-use disorders in DSM-III-R. Evidence for the dependence syndrome across different psychoactive substances. Br J Psychiatry. 1987 Dec;151:834–43. PubMed
- Yehuda R, Southwick SM, Giller EL. Exposure to atrocities and severity of chronic posttraumatic stress disorder in Vietnam combat veterans. Am J Psychiatry. 1992;149(3):333–336. PubMed CrossRef
- Yehuda R, Southwick S, Giller EL, et al. Urinary catecholamine excretion and severity of PTSD symptoms in Vietnam combat veterans. J Nerv Ment Dis. 1992 May;180(5):321–5. PubMed
- Liberzon I, Taylor SF, Amdur R, et al. Brain activation in PTSD in response to trauma-related stimuli. Biol Psychiatry. 1999 Apr 1;45(7):817–26. PubMed
- Liberzon I, Abelson JL, Flagel SB, et al. Neuroendocrine and psychophysiologic responses in PTSD: a symptom provocation study. Neuropsychopharmacology. 1999 Jul;21(1):40–50. PubMed
- Geracioti TD Jr, Baker DG, Ekhator NN, et al. CSF norepinephrine concentrations in posttraumatic stress disorder. Am J Psychiatry. 2001 Aug;158(8):1227–30. PubMed
- Strawn JR, Geracioti TD Jr. Noradrenergic dysfunction and the psychopharmacology of posttraumatic stress disorder. Depress Anxiety. 2008;25(3):260–71. PubMed
- McCall A, Forouhandehpour R, Celebi S, et al. Evidence for locus coeruleus-norepinephrine system abnormality in military posttraumatic stress disorder revealed by neuromelanin-sensitive magnetic resonance imaging. Biol Psychiatry. 2024 Aug 15;96(4):268–277. PubMed
- Kolb LC, Burris BC, Griffiths S. Propranolol and clonidine in the treatment of the chronic post-traumatic stress disorders of war. In: Kolk BA, ed. Post-traumatic Stress Disorder: Psychological and Biological Sequelae. American Psychiatric Press; 1984:98–105.
- Kinzie JD, Leung P. Clonidine in Cambodian patients with posttraumatic stress disorder. J Nerv Ment Dis. 1989 Sep;177(9):546–50. PubMed
- Neylan TC, Lenoci M, Samuelson KW, et al. No improvement of posttraumatic stress disorder symptoms with guanfacine treatment. Am J Psychiatry. 2006 Dec;163(12):2186–8. PubMed
- Davis LL, Ward C, Rasmusson A, et al. A placebo-controlled trial of guanfacine for the treatment of posttraumatic stress disorder in veterans. Psychopharmacol Bull. 2008;41(1):8–18. PubMed
- Raskind MA, Peskind ER, Hoff DJ, et al. A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Biol Psychiatry. 2007 Apr 15;61(8):928–34. PubMed
- Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013 Sep;170(9):1003–10. PubMed
- Raskind MA, Peskind ER, Chow B, et al. Trial of prazosin for post-traumatic stress disorder in military veterans. N Engl J Med. 2018 Feb 8;378(6):507–517. PubMed
- Germain A, Richardson R, Moul DE, et al. Placebo-controlled comparison of prazosin and cognitive-behavioral treatments for sleep disturbances in US Military Veterans. J Psychosom Res. 2012 Feb;72(2):89–96. PubMed
- Villarreal G, Hamner MB, Cañive JM, et al. Efficacy of quetiapine monotherapy in posttraumatic stress disorder: a randomized, placebo-controlled trial. Am J Psychiatry. 2016 Dec 1;173(12):1205–1212. PubMed
- O’Connor M. Adjunctive therapy with brexpiprazole improves treatment resistant complex post traumatic stress disorder in domestic family violence victims. Australas Psychiatry. 2020 Jun;28(3):264–266. PubMed
- Swearer JM, Drachman DA, O’Donnell BF, et al. Troublesome and disruptive behaviors in dementia. Relationships to diagnosis and disease severity. J Am Geriatr Soc. 1988 Sep;36(9):784–90. PubMed CrossRef
- Steele C, Rovner B, Chase GA, et al. Psychiatric symptoms and nursing home placement of patients with Alzheimer’s disease. Am J Psychiatry. 1990 Aug;147(8):1049–51. PubMed CrossRef
- Raskind MA, Peskind ER, Halter JB, et al. Norepinephrine and MHPG levels in CSF and plasma in Alzheimer’s disease. Arch Gen Psychiatry. 1984 Apr;41(4):343–6. CrossRef
- Elrod R, Peskind ER, DiGiacomo L, et al. Effects of Alzheimer’s disease severity on cerebrospinal fluid norepinephrine concentration. Am J Psychiatry. 1997 Jan;154(1):25–30. CrossRef
- Hoogendijk WJ, Feenstra MG, Botterblom MH, et al. Increased activity of surviving locus ceruleus neurons in Alzheimer’s disease. Ann Neurol. 1999 Jan;45(1):82–91. CrossRef
- Szot P, White SS, Greenup JL, et al. Compensatory changes in the noradrenergic nervous system in the locus ceruleus and hippocampus of postmortem subjects with Alzheimer’s disease and dementia with Lewy bodies. J Neurosci. 2006 Jan 11;26(2):467–78. CrossRef
- Sharp SI, Ballard CG, Chen CP, Francis PT. Aggressive behavior and neuroleptic medication are associated with increased number of alpha1-adrenoceptors in patients with Alzheimer disease. Am J Geriatr Psychiatry. 2007 May;15(5):435–7. CrossRef
- Peskind ER, Wingerson D, Murray S, et al. Effects of Alzheimer’s disease and normal aging on cerebrospinal fluid norepinephrine responses to yohimbine and clonidine. Arch Gen Psychiatry. 1995 Sep;52(9):774–82. CrossRef
- Peskind ER, Tsuang DW, Bonner LT, et al. Propranolol for disruptive behaviors in nursing home residents with probable or possible Alzheimer disease: a placebo-controlled study. Alzheimer Dis Assoc Disord. 2005 Jan-Mar;19(1):23–8. CrossRef
- Wang LY, Shofer JB, Rohde K, et al. Prazosin for the treatment of behavioral symptoms in patients with Alzheimer disease with agitation and aggression. Am J Geriatr Psychiatry. 2009 Sep;17(9):744–51. CrossRef
- Rabinowitz J, Harvey PD, Eerdekens M, et al. Premorbid functioning and treatment response in recent-onset schizophrenia. Br J Psychiatry. 2006 Jul;189:31–5. CrossRef
- Lee D, Slomkowski M, Hefting N, et al. Brexpiprazole for the treatment of agitation in Alzheimer dementia: a randomized clinical trial. JAMA Neurol. 2023 Dec 1;80(12):1307–1316. CrossRef
- Almulla AF, Al-Hakeim HK, Maes M. Schizophrenia phenomenology revisited: Positive and negative symptoms are strongly related reflective manifestations of an underlying single trait indicating overall severity of schizophrenia. CNS Spectr. 2021;26(4):368–377. CrossRef
- Borsboom D. A network theory of mental disorders. World Psychiatry. 2017;16(1):5–13. CrossRef
- Cramer AO, Waldorp LJ, van der Maas HL, et al. Comorbidity: A network perspective. Behav Brain Sci. 2010;33(2–3):137–150; discussion 150–193. CrossRef
- Menon V. Large-scale brain networks and psychopathology: A unifying triple network model. Trends Cogn Sci. 2011;15(10):483–506. CrossRef
- Schimmelpfennig J, Topczewski J, Zajkowski W, et al. The role of the salience network in cognitive and affective deficits. Front Hum Neurosci. 2023;17:1133367. CrossRef
- Zerbi V, Floriou-Servou A, Markicevic M, et al. Rapid reconfiguration of the functional connectome after chemogenetic locus coeruleus activation. Neuron. 2019;103(4):702–718.e705. CrossRef
- Lee D, Slomkowski M, Hefting N, et al. Brexpiprazole for the treatment of agitation in Alzheimer dementia: A randomized clinical trial. JAMA Neurol. 2023;80(12):1307–1316. CrossRef
- Mehta R, Bhattacharya R, Mallick BN. Sleep and neuroimmunomodulation for maintenance of optimum brain function: Role of noradrenaline. Brain Sci. 2022;12(12). CrossRef
- Hillhouse TM, Porter JH. A brief history of the development of antidepressant drugs: From monoamines to glutamate. Exp Clin Psychopharmacol. 2015;23(1):1–21. CrossRef
- Hu X, Pan L, Li W. Meta-analysis on the efficacy of the norepinephrine reuptake inhibitors reboxetine and atomoxetine for the treatment of schizophrenia and attention deficit hyperactivity disorder. Adv Clin Exp Med. 2023;32(5):511–522. CrossRef
This PDF is free for all visitors!