ABSTRACT
Objective: Determine if sublingual dexmedetomidine, a selective α2 adrenergic receptor agonist, reduces symptoms of acute agitation associated with schizophrenia or schizoaffective disorder.
Methods: This phase 3, randomized, double-blind, placebo-controlled study was conducted in adults diagnosed with schizophrenia or schizoaffective disorder per the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) criteria. The study was conducted at 15 US sites between January 23, 2020, and May 8, 2020. Participants were randomized to sublingual dexmedetomidine 180 μg, 120 μg, or matching placebo. The primary efficacy endpoint was mean change from baseline in the Positive and Negative Syndrome Scale-Excited Component (PEC) total score at 2 hours postdose.
Results: Altogether, 380 participants (mean age 45.6 years, 63.4% identifying as male, 77.9% identifying as Black or African American) were randomized; 380 (100%) self-administered study medication, and 372 (97.9%) completed the study. The mean PEC total score at baseline (17.6) indicated mild to moderate agitation. At 2 hours postdose, the least squares mean changes (SE) from baseline were −10.3 (0.4) for sublingual dexmedetomidine 180 μg, −8.5 (0.4) for 120 μg, and −4.8 (0.4) for placebo. Least squares mean differences (97.5% confidence intervals) in the sublingual dexmedetomidine groups were −5.5 (−6.7 to −4.3) for 180 μg and −3.7 (−4.9 to −2.5) for 120 μg (both P < .001 vs placebo). The most commonly encountered adverse events with dexmedetomidine (incidence ≥ 5% and ≥ 2× rate observed with placebo) were somnolence, dry mouth, and hypotension for the 120 μg dose, and somnolence, dizziness, orthostatic hypotension, and oral hypoesthesia for the 180 μg dose.
Conclusions: Treatment with sublingual dexmedetomidine 180 μg or 120 μg was more efficacious than placebo in reducing acute agitation associated with schizophrenia as measured by PEC scores at 2 hours postdose.
Trial Registration: ClinicalTrials.gov identifier: NCT04268303
J Clin Psychiatry 2022;83(6):22m14447
To cite: Citrome L, Preskorn SH, Lauriello J, et al. Sublingual dexmedetomidine for the treatment of acute agitation in adults with schizophrenia or schizoaffective disorder: a randomized placebo-controlled trial. J Clin Psychiatry. 2022;83(6):22m14447.
To share: https://doi.org/10.4088/JCP.22m14447
© 2022 Physicians Postgraduate Press, Inc.
aDepartment of Psychiatry and Behavioral Sciences, New York Medical College, Valhalla, New York
bKansas University School of Medicine-Wichita, Wichita, Kansas
cDepartment of Psychiatry and Human Behavior, Thomas Jefferson University, Philadelphia, Pennsylvania
dDepartment of Psychiatry, Yale University, New Haven, Connecticut
eSegal Trials, Fort Lauderdale, Florida
fJupiter Point Pharma Consulting, LLC, Groton, Connecticut
gBioXcel Therapeutics, Inc., New Haven, Connecticut
*Corresponding author: Leslie Citrome, MD, MPH, 11 Medical Park Drive, Ste 102, Pomona, NY 10970 ([email protected]).
Agitation is a syndrome encompassing symptoms of restlessness, irritability, anxiety, and increased or excessive movement or speech that lacks specific purpose and that can escalate to aggressive or violent behavior.1–3 In many individuals, agitation emerges when patients feel anxious, angry, or threatened, or when their ability to resolve their distress is compromised, as might occur in an unfamiliar context or during a state of intoxication.2 Schizophrenia is among the most common mental health conditions associated with agitation. In one multicenter observational study in 27 European psychiatric emergency departments and acute inpatient units, approximately 47% of all episodes of agitation during the observation period were associated with a diagnosis of schizophrenia.4 Evidence suggests that patients with schizophrenia had difficulty self-managing their symptoms of agitation,5 which can lead to violent behavior, disruption in health care settings, and injury to patients and staff. As a result, episodes of agitation in patients with schizophrenia warrant immediate attention and medical care.3,6–8
The recommended initial treatment of acute agitation involves nonpharmacological methods, such as verbal de-escalation and environment management.9 If response is inadequate and medication is required, antipsychotics and benzodiazepines are commonly administered.10 In the event of an escalation, there is an increased probability that physical restraint and seclusion will be required with an associated need for increased staffing resources and longer duration of stay.2,11 The importance of avoiding physical restraint is highlighted in 2 recent reviews describing evidence for psychological and physical harm resulting from restraint.12,13 There remains a need for a rapidly effective, noninvasive treatment for acute agitation with a favorable safety and tolerability profile. Until now, all US Food and Drug Administration (FDA)-approved agents are administered intramuscularly14 or in the deep lung through an inhalation device.15
Approved by the FDA on April 5, 2022, dexmedetomidine sublingual film (IGALMI, manufactured and distributed by BioXcel Therapeutics, Inc., New Haven, CT) is indicated in adults for the acute treatment of agitation associated with schizophrenia or bipolar I or II disorder.16 Dexmedetomidine is a well-known efficacious α2-adrenergic receptor agonist available since 1999 in an intravenous formulation indicated for sedation of initially intubated and mechanically ventilated patients in an intensive care unit setting and sedation of non-intubated patients prior to and/or during surgical and other procedures.17 The reformulation of dexmedetomidine as a sublingual film allows the broader use of this agent in psychiatric settings when managing agitation, thus potentially avoiding the use of intramuscular administration of antipsychotics and/or benzodiazepines. Non-invasive formulations, although requiring cooperation from patients, have the potential to improve overall patient experience, thereby improving future cooperation between patients and health care providers.3 The sublingual film formulation is orally absorbed, thus avoiding first-pass hepatic metabolism and providing higher dexmedetomidine bioavailability than ingested formulations.18,19 In previous research, this formulation demonstrated dose-dependent exposure and a plasma half-life between 2 and 3 hours.18 The absolute bioavailability was approximately 72% and 82% following sublingual and buccal administration, respectively. The objective of the present study was to determine if a single dose of sublingual dexmedetomidine effectively reduces symptoms of acute agitation associated with schizophrenia or schizoaffective disorder. A similarly designed study demonstrated efficacy in acute agitation associated with bipolar disorder.20
METHODS
This study was conducted in accordance with the principles of the Guidelines for Good Clinical Practice, the Declaration of Helsinki, and all applicable local regulations. The protocol was approved by independent ethics committees and/or institutional review boards at each study center and by a central institutional review board (Advarra, Inc.). Participants provided written informed consent before any study procedures were undertaken. The study was prospectively registered at ClinicalTrials.gov (identifier: NCT04268303).
Design
This phase 3, randomized, double-blind, placebo-controlled study was conducted at 15 hospitals and research units in the United States between January 23, 2020, and May 8, 2020, to evaluate the safety and efficacy of sublingual dexmedetomidine for the treatment of acute agitation in adults with schizophrenia or schizoaffective disorder. Although patients with schizophreniform disorder were also eligible to participate, there were no subjects who met diagnostic criteria for schizophreniform disorder. Participants were assessed at screening, treatment (day 1), follow-up (day 2), discharge (day 3), and end of study (day 7). Participants were housed in a clinical research setting or hospitalized under medical supervision. They remained in the clinical unit until at least the morning of day 3, the earliest day of discharge.
Participants
Eligible participants were aged 18–75 years with a diagnosis of schizophrenia or schizoaffective disorder, according to the criteria of the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition,21 as assessed by the Mini-International Neuropsychiatric Interview (MINI).22 They had presented with acute agitation in outpatient clinics or mental health, psychiatric, or medical emergency services (including medical/psychiatric observation units) or as inpatients newly admitted for acute agitation or already hospitalized for underlying conditions. Participants were required to have a total score of ≥ 14 on the 5-item Positive and Negative Syndrome Scale-Excited Component (PEC)23 scale at screening and baseline and a score of ≥ 4 on at least 1 PEC item at baseline. Participants provided informed consent prior to enrollment and had to be capable and willing to self-administer study medication.
Treatments
Sublingual dexmedetomidine 180 μg, sublingual dexmedetomidine 120 μg, and placebo were provided as individual films in a heat-sealed white foil pouch. While the film containing active drug appeared subtly different than a placebo film, participants were unaware of any visual difference, having never previously seen an active or placebo film. Participants were instructed on the appropriate method of self-administration, and study drug was self-administered under the supervision of a staff member. At investigator discretion in the event of persistent or recurrent agitation, a second dose of sublingual dexmedetomidine 90 μg or 60 μg (half of the 180 μg or 120 μg initial dose) or placebo could be given 2 hours after the first dose rather than rescue medication, if the change from baseline on the PEC scale was less than 40% and if there were no safety concerns (eg, hypotension, sedation). Participants with inadequate relief of agitation after a second sublingual dexmedetomidine dose could be given, at investigator discretion and in lieu of rescue medication, a third dose of sublingual dexmedetomidine or placebo. The maximum number of study medication doses permitted was 3. At the discretion of the principal investigator, if the patient’s status warranted, standard of care rescue treatment for agitation with lorazepam 0.5 to 5 mg oral or intramuscular could be initiated at any time.
Outcomes
The primary efficacy endpoint was the absolute change from baseline in the PEC total score at 2 hours postdose. The PEC scale includes 5 items—poor impulse control, tension, hostility, uncooperativeness, and excitement—each of which is rated from 1 (minimum) to 7 (maximum); the sum of these 5 items, the PEC total score, ranges from 5 (absence of agitation) to 35 (extremely severe). The PEC was administered at screening, predose (within 15 minutes prior to dose), and at 10, 20, 30, 45, 60, 90 minutes and 2, 4, 6, 8, and 24 hours postdose. At all timepoints, the PEC was administered before any other assessments.
Secondary endpoints included the absolute change from baseline in the PEC total score at 10, 20, 30, 45, 60, and 90 minutes postdose.
Multiple exploratory endpoints were prespecified. The Clinical Global Impression-Improvement (CGI-I) scale,24 with possible scores ranging from 1 (very much improved) to 7 (very much worse), was used to rate overall clinical improvement at 30 minutes, 1, 2, and 4 hours postdose. The Agitation-Calmness Evaluation Scale (ACES) is a single-item measure (ranging from 1 = marked agitation to 9 = unarousable) that was used to assess overall agitation predose (within 15 minutes of dosing) and 2, 4, and 8 hours postdose. The PEC response rate (≥ 40% reduction in total score from baseline at or before 2 hours postdose)23 and change from baseline in total PEC score from 10 minutes through 24 hours postdose were assessed. The CGI-I response rate, defined as a score of 1 or 2 (very much or much improved) at 2 hours postdose was also assessed. Agitation/calmness was further assessed by measuring change from baseline for PEC total score at 4, 6, 8, and 24 hours postdose.
The safety and tolerability of sublingual dexmedetomidine were evaluated based on adverse events (AEs); laboratory tests (chemistry, hematology, urinalysis); electrocardiography; pulse oximetry; and vital signs (systolic blood pressure, diastolic blood pressure, and heart rate). Adverse events were characterized by type, severity, seriousness, and relationship to treatment and coded by preferred term and system organ class using the Medical Dictionary for Regulatory Activities, version 23.0. A buccal examination for local irritation was performed at 30 minutes and 2, 4, and 24 hours postdose. The Columbia-Suicide Severity Rating Scale (C-SSRS) was performed at screening, baseline, 24 hours postdose, and discharge.25
All reported AEs were recorded by the site investigators and assessed for severity per protocol using the following definitions:
Mild: Is usually transient and may require only minimal treatment or therapeutic intervention. The event does not generally interfere with usual activities of daily living.
Moderate: Is usually alleviated with additional specific therapeutic intervention. The event interferes with usual activities of daily living, causing discomfort, but poses no significant or permanent risk of harm to the subject.
Severe: Interrupts usual activities of daily living, significantly affects clinical status, or may require intensive therapeutic intervention.
Sample Size Calculation
Sample size was calculated based on the results of a phase 2 study of sublingual dexmedetomidine. For the analysis of the primary efficacy endpoint, it was estimated that 125 participants per treatment group would be needed to detect at least a 2.1-unit change from baseline in the PEC total score for the sublingual dexmedetomidine-placebo pairwise comparison (1:1:1 randomization, 2-tailed α = .025, power = 90%).
Randomization
Participants were randomized (1:1:1) to sublingual dexmedetomidine 180 μg, sublingual dexmedetomidine 120 μg, or matching placebo film and stratified by age (< 65, ≥ 65 years). Randomization was computer generated by permuted block design with a block size of 6. Participants, investigators, and study staff were blinded to the identity of the assigned treatment. The active treatment and the placebo were similar in taste and lightly mint-flavored.
Statistical Analysis
Analyses were conducted using SAS/STAT software, version 9.4 (SAS Institute Inc, Cary, NC).
The safety population included all participants who received a dose of study drug. The efficacy (intent-to-treat) population included all participants in the safety population who had at least 1 PEC assessment postdose.
The null and alternate hypotheses for efficacy were tested using a mixed model repeated measures (MMRM) analysis. To control for type 1 error associated with multiple comparisons of sublingual dexmedetomidine with placebo, the 2-tailed α was set at 0.025, and Bonferroni correction was applied. For the MMRM analysis, the change from baseline in the PEC score at 10, 20, 30, 45, 60, and 90 minutes and 2 hours postdose was the outcome variable; covariates included treatment group, baseline PEC, visit, baseline PEC-by-visit interaction term, age stratum, study site, and treatment group–by-visit interaction term. The difference in the mean change from baseline in each sublingual dexmedetomidine group relative to placebo, as well as the significance levels associated with the null hypotheses, were obtained from differences in least squares means at each time point.
To control for multiplicity on secondary endpoints, a hierarchical testing procedure and a prespecified sequence of comparisons were used; starting at 90 minutes postdose, each earlier endpoint was tested in succession until a comparison with placebo failed to achieve statistical significance at the .025 level. Endpoints following a nonsignificant result in the hierarchy were not formally tested.
As comparisons on exploratory endpoints were not controlled for multiplicity, the results should be considered hypothesis-generating.
There were no missing values in the primary or secondary outcome data. For the efficacy analyses, all values collected after the use of rescue treatment or withdrawal from the study were censored, and these participants were considered nonresponders.
RESULTS
Participants
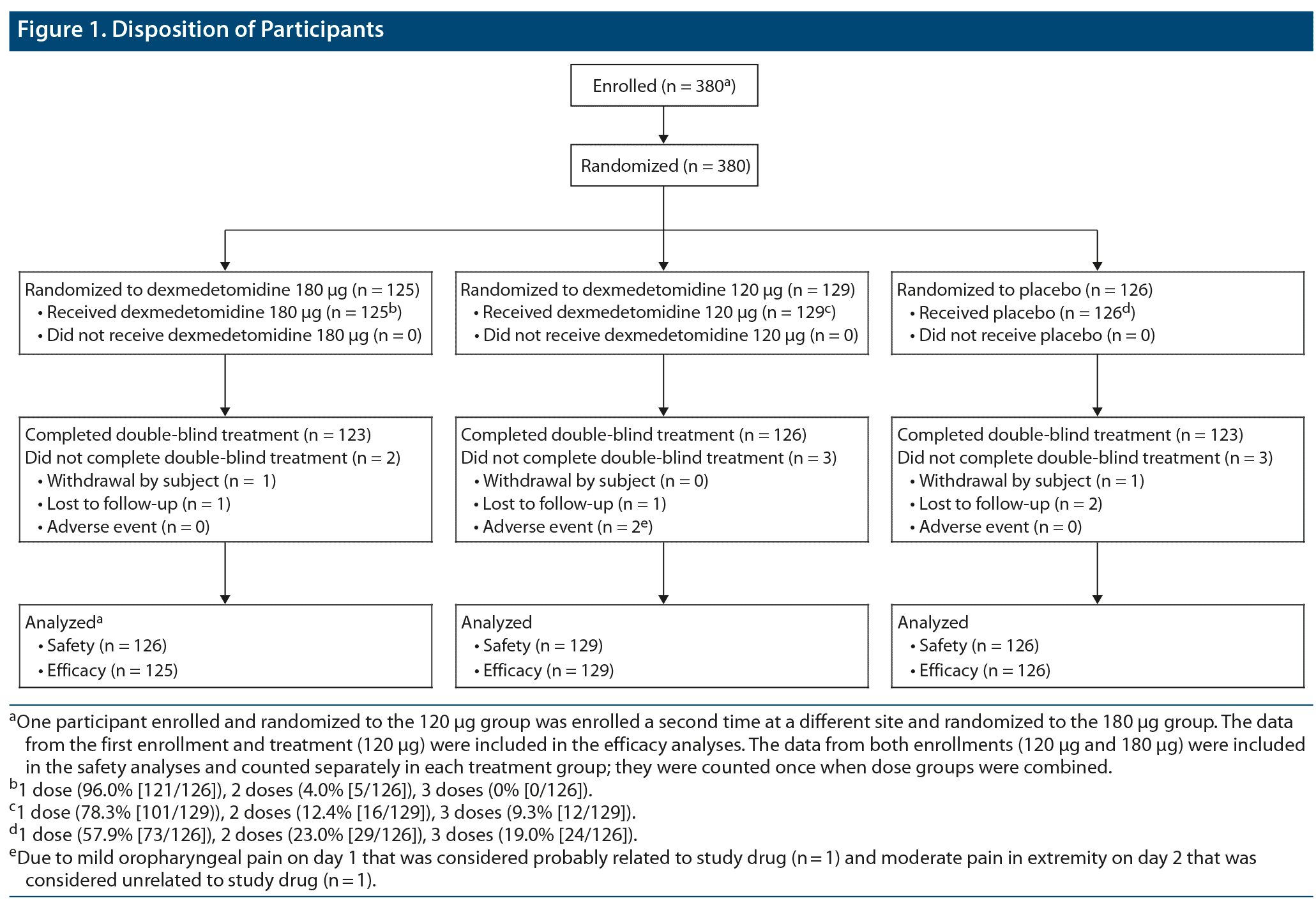
Three hundred eighty participants were randomized to sublingual dexmedetomidine 180 μg (n = 125), sublingual dexmedetomidine 120 μg (n = 129), or placebo (n = 126) and analyzed for efficacy; of these, 372 (97.9%) completed the study (Figure 1).
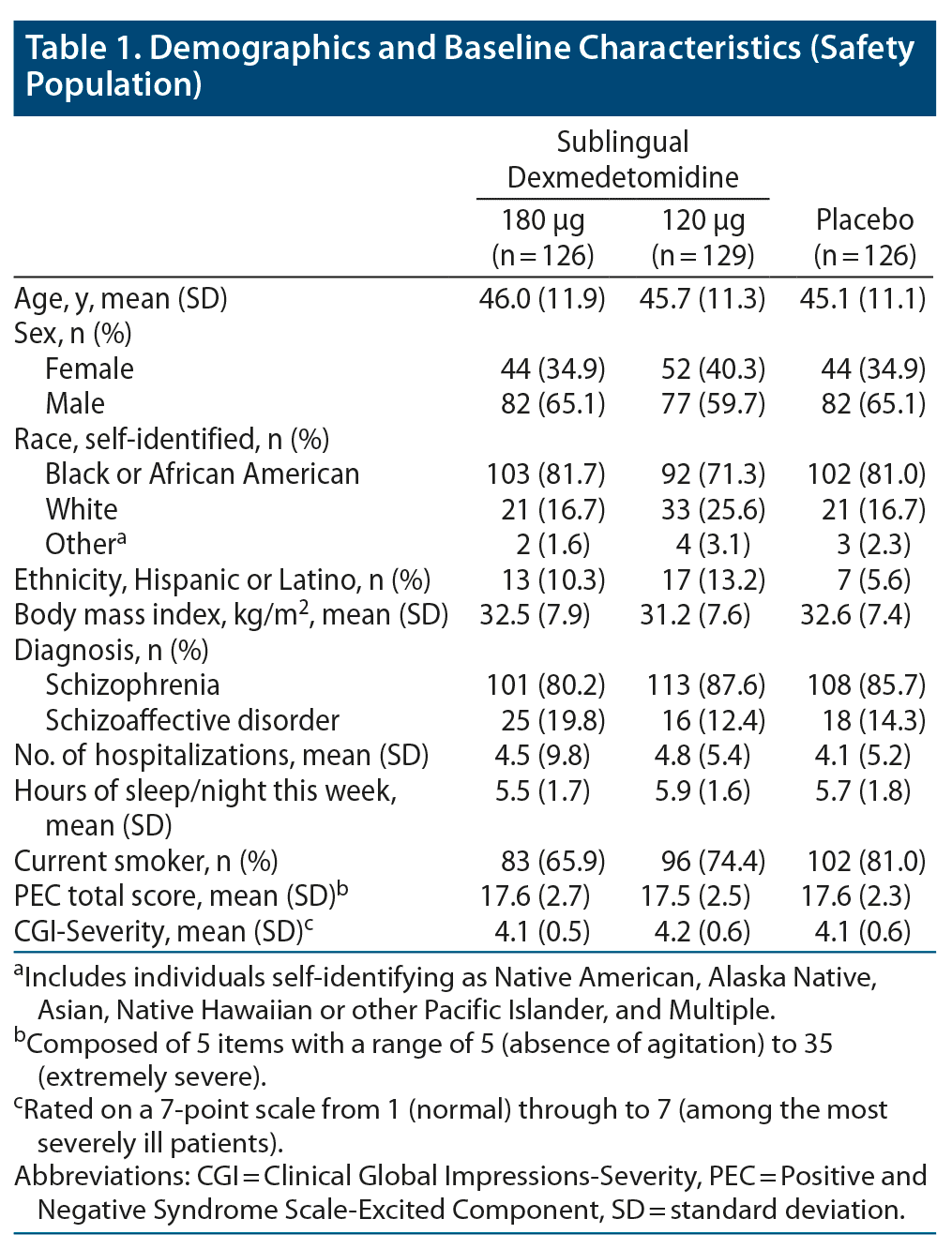
The treatment groups were generally comparable at baseline (Table 1). The population had a mean (SD) age of 45.6 (11.4) years; 63.4% (241/380) of participants identified as male, and 77.9% identified as Black or African American, which reflects the demographics of the population served by the study sites. The mean (SD) body mass index was 32.1 (7.6) kg/m2. Diagnoses at screening were schizophrenia (84.5% [321/380]) and schizoaffective disorder (15.5% [59/380]). Seven participants were aged 65 years or older (2 in the sublingual dexmedetomidine 180 μg group, 3 in the sublingual dexmedetomidine 120 μg group, and 2 in the placebo group). Concomitant psychiatric medications are listed in Supplementary Table 1.
One participant was enrolled and randomized twice, once to sublingual dexmedetomidine 120 μg and once (at a different site) to sublingual dexmedetomidine 180 μg. Data from the first enrollment and treatment (120 μg) were included in the efficacy analyses, and data from both enrollments (120 μg and 180 μg) were included in the safety analyses. In total, 2.1% (8/380) of participants discontinued; 0.5% (2/380) voluntarily withdrew (1 participant each in the sublingual dexmedetomidine 180 μg and placebo groups), 0.5% (2/380) in the sublingual dexmedetomidine 120 μg group discontinued due to AEs of mild oropharyngeal pain on day 1 and moderate pain in extremity on day 7, and 1.1% were lost to follow-up (1 participant in each active treatment group and 2 in the placebo group).
Two doses of study medication were administered to 4.0% (5/126) of participants in the sublingual dexmedetomidine 180 μg group, 12.4% (16/129) in the 120 μg group, and 23.0% (29/126) in the placebo group. No participant in the 180 μg group received 3 doses of study medication; however, 9.3% (12/129) of participants in the 120 μg group and 19.0% (24/126) in the placebo group received a total of 3 doses of study medication (Supplementary Table 2). Rates of rescue medication use for persistent agitation were 0.8% (1/125) in the sublingual dexmedetomidine 180 μg group, 3.1% (4/129) in the sublingual dexmedetomidine 120 μg and, 0.8% (1/126) in the placebo group. No participant received both rescue medication and more than 1 dose of study medication.
Efficacy
At 2 hours postdose (Figure 2), the least squares mean (standard error [SE]) changes from baseline in PEC total score were −10.3 (0.4) for sublingual dexmedetomidine 180 μg, −8.5 (0.4) for sublingual dexmedetomidine 120 μg, and −4.8 (0.4) for placebo. Least squares mean (97.5% confidence interval [CI]) differences from placebo at 2 hours postdose were −5.5 (−6.7 to −4.3, P < .001) for sublingual dexmedetomidine 180 μg and −3.7 (−4.9 to −2.5, P < .001) for sublingual dexmedetomidine 120 μg.
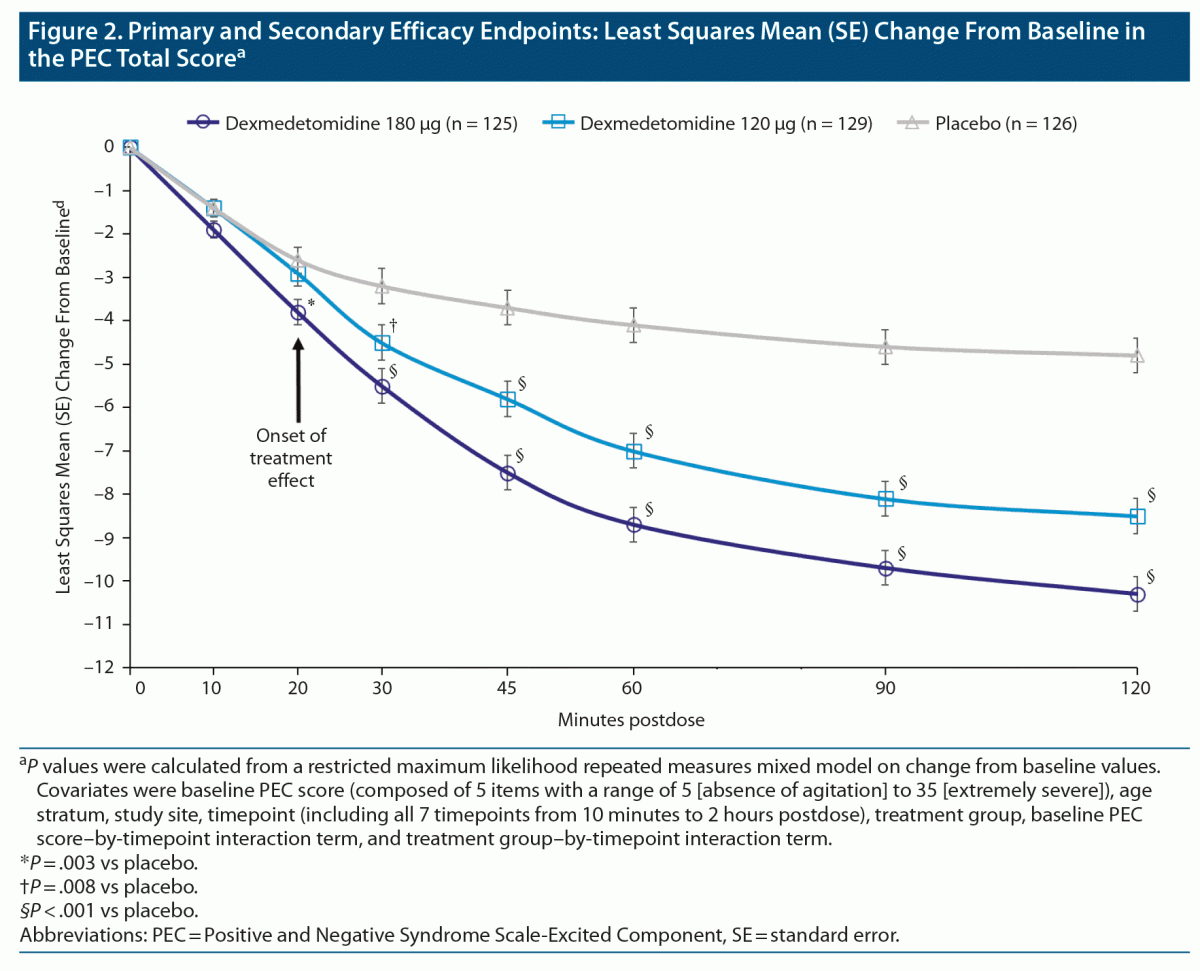
Treatment effects were first observed at 20 minutes postdose with sublingual dexmedetomidine 180 μg (least squares mean difference, standard error [97.5% CI]: −1.2, 0.4 [−2.1 to −0.3], P = .003]) and at 30 minutes postdose with sublingual dexmedetomidine 120 μg (least squares mean difference, SE [97.5% CI]: −1.3, 0.5 [−2.4 to −0.2], P = .008). Participants in both sublingual dexmedetomidine treatment groups showed improvements compared with participants in the placebo group at all subsequent timepoints (45, 60, and 90 minutes postdose (P < .001; Figure 2).
Results on the exploratory endpoints—response on the PEC total score, change from baseline and response on the CGI-I, and change from baseline and resolution of agitation on the ACES—are presented in Supplementary Box 1, Supplementary Table 3, and Supplementary Figures 1–4.
Safety
The incidence of AEs was 37.3% (47/126) with sublingual dexmedetomidine 180 μg, 39.5% (51/129) with sublingual dexmedetomidine 120 μg, and 15.1% (19/126) with placebo (Table 2). There were no reports of severe or serious AEs. Two (1.7%) participants in the sublingual dexmedetomidine 120 μg group discontinued due to AEs; 1 experienced pain in extremity (moderate severity and judged not related to study drug by investigator) and the other experienced oropharyngeal pain (mild severity and judged probably related to study drug by investigator). The most common AE in both sublingual dexmedetomidine treatment groups was somnolence (180 μg: 23.0% [29/126], 120 μg: 21.7% [28/129], placebo: 7.9% [10/126]), which was mild in 82.8% of the 180 μg group, 89.3% of the 120 μg group, and 100.0% of the placebo group. Other common AEs in the sublingual dexmedetomidine treatment groups included headache (3.2%–4.7%), hypoesthesia (3.9%–5.6%), hypotension and orthostatic hypotension (1.6%–6.2%), and dry mouth (4.0%–7.8%).
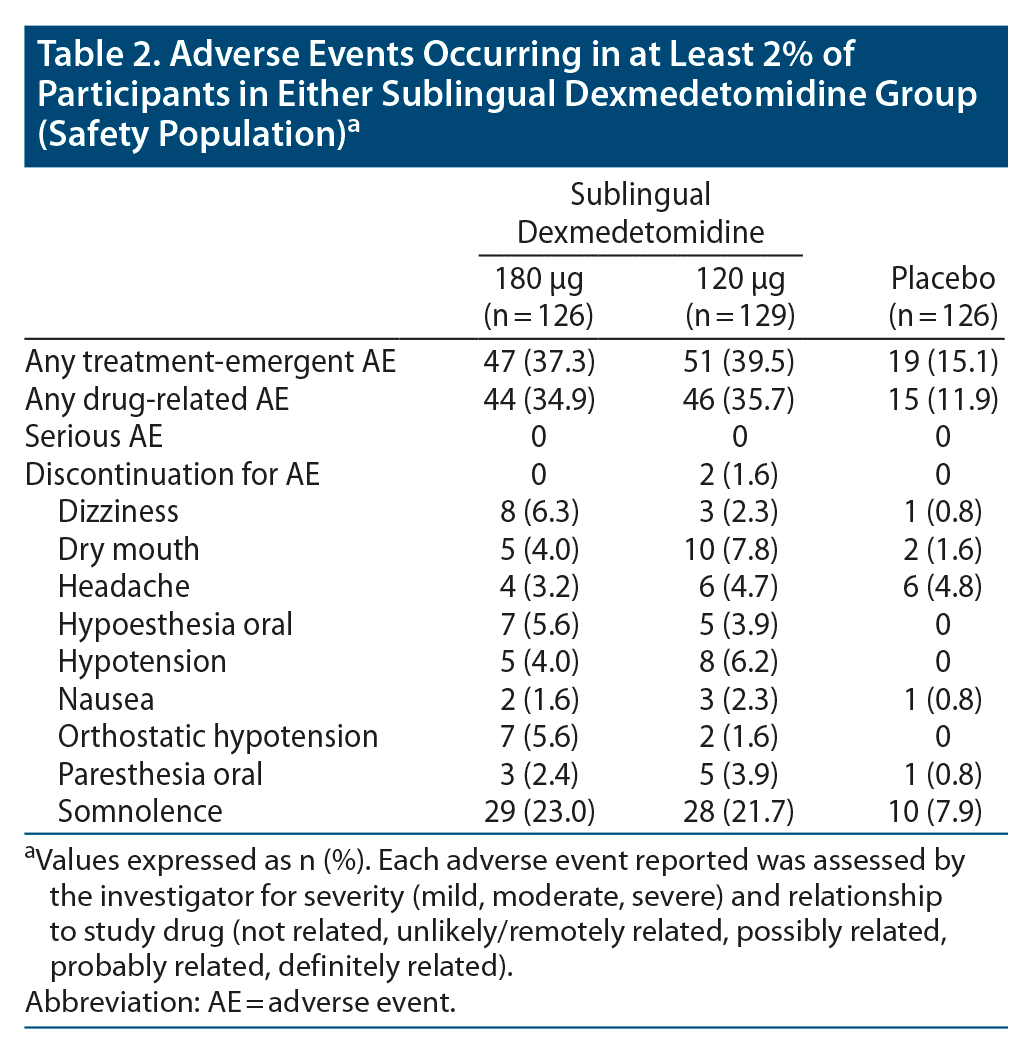
No clinically meaningful changes in clinical laboratory values or physical examination findings were observed. There were no clinically meaningful changes from baseline at 2 hours or 24 hours postdose for PR interval, QRS duration, or QTcF. All AEs of hypotension, orthostatic hypotension, and bradycardia were reviewed by an external board-certified cardiologist. An AE of hypotension occurred in 5 (4.0%) participants in the sublingual dexmedetomidine 180 μg group and 8 (6.2%) participants in the 120 μg group; the events were judged by the investigator as mild in 9 and moderate in 4 participants, and all resolved without medical intervention. When assessed by an independent external cardiologist, 2 of the 5 cases of hypotension in the 180 μg group and 3 of the 8 cases in the 120 μg group were considered to be not clinically meaningful (ie, not associated with abnormal vital sign measurements).
No participant in the placebo group experienced an AE of hypotension. Seven (5.6%) participants in the sublingual dexmedetomidine 180 μg group, 2 (1.6%) participants in the sublingual dexmedetomidine 120 μg group, and no (0%) participants in the placebo group experienced an AE of orthostatic hypotension. Two (1.6%) participants in the sublingual dexmedetomidine 120 μg group experienced an AE of sinus bradycardia, and both cases resolved without medical intervention.
At 2 hours postdose, participants treated with sublingual dexmedetomidine had reductions in systolic blood pressure (180 μg: −16.8 [14.8] mm Hg, 120 μg: −12.8, [13.7] mm Hg); diastolic blood pressure (180 μg: −8.9, [10.2] mm Hg, 120 μg: −7.7, [8.5] mm Hg); and heart rate (180 μg: −8.2, [10.3] bpm, 120 μg: −7.6, [9.4]). Similar reductions in blood pressure were not observed in placebo-treated participants. One (0.8%) participant in the sublingual dexmedetomidine 120 μg group experienced suicidal ideation as assessed by the C-SSRS on day 2 but not on day 3; there was no suicidal behavior, and the participant subsequently completed the study. On buccal examination, 1 (0.8%) participant in the sublingual dexmedetomidine 180 μg group experienced local irritation at 4 hours postdose that was resolved by 24 hours postdose.
DISCUSSION
This phase 3, randomized, double-blind, placebo-controlled study was conducted to determine if sublingual dexmedetomidine, a selective α2-adrenergic receptor agonist, reduces symptoms of acute agitation associated with schizophrenia. Results showed that a single dose of 180 μg or 120 μg sublingual dexmedetomidine reduced the severity of agitation in participants with schizophrenia or schizoaffective disorder as assessed by absolute change from baseline in PEC score. Sublingual dexmedetomidine produced improvement compared with placebo at 2 hours postdose, and treatment effects were evident within 20 minutes postdose among participants who received sublingual dexmedetomidine 180 μg and within 30 minutes postdose among those who received sublingual dexmedetomidine 120 μg. Repeat dosing was more frequent in the placebo group than in either sublingual dexmedetomidine group. More participants in the sublingual dexmedetomidine groups than in the placebo group experienced AEs of somnolence, but most events were mild or moderate, and no participant was unarousable based on AE report or on ACES rating scale score.
The observed effect sizes on the primary outcome measure in the sublingual dexmedetomidine groups in this study were similar to those reported in registration trials of intramuscularly administered agents indicated for the treatment of agitation associated with schizophrenia, and participants had baseline agitation of comparable severity.14 Given the potential for benefit in this patient population, the positive results with sublingual dexmedetomidine on multiple prospectively defined exploratory endpoints, together with its tolerability and safety profiles, merit further evaluation. Moreover, unlike the antipsychotics commonly used to manage agitation, dexmedetomidine does not impact on dopamine receptors, and thus motoric adverse events such as dystonia, tremor, and akathisia are avoided.
Despite its strengths, this study has some limitations. First, efficacy and tolerability were assessed following a single episode of agitation, providing no information on the efficacy and safety of longer-term use of dexmedetomidine. Second, the controlled nature of a clinical trial and capacity of participants to undergo informed consent prior to enrollment may limit the generalizability of these results to individuals who present with acute agitation in emergency settings. Third, participants were excluded for acute intoxication but were not screened for possible alcohol withdrawal that may have contributed to an agitated state. Fourth, participants could have received antipsychotic medications prior to study entry that may have contributed to akathisia or restlessness. Fifth, 78% of the participants in the study self-identified as Black or African American, which reflects the demographics of the population served by the study sites. Finally, rates of rescue medication use were likely reduced by the design of the protocol that allowed investigators to use study drug instead of per-protocol rescue medication (lorazepam) for participants with inadequate relief of agitation at 2 hours postdose and beyond.
Guidance for pharmacologic treatment of acute agitation in psychiatric conditions includes administration of antipsychotics and/or benzodiazepines.2,11,26 These consensus recommendations state that, ideally, a drug should have an onset of action of ≤ 30 minutes, induce calmness, provide ease of administration, and be noninvasive, nontraumatic, safe, and well tolerated.2,27 In light of these recommendations, the results of the present study suggest that sublingual dexmedetomidine may be a useful addition to the armamentarium in the treatment of agitation associated with schizophrenia or schizoaffective disorder.
CONCLUSION
Among participants with mild-to-moderate agitation associated with schizophrenia or schizoaffective disorder, treatment with sublingual dexmedetomidine 120 μg or 180 μg resulted in greater reduction in agitation, as measured by the PEC score at 2 hours postdose, compared with placebo.
Submitted: March 8, 2022; accepted May 23, 2022.
Published online: October 3, 2022.
Relevant financial relationships: Dr Citrome reported serving as chair of the drug monitoring committee for this study and as consultant to AbbVie/Allergan, Acadia, Adamas, Alkermes, Angelini, Astellas, Avanir, Axsome, BioXcel, Boehringer Ingelheim, Cadent Therapeutics, Eisai, Enteris BioPharma, HLS Therapeutics, Impel, Intra-Cellular Therapies, Janssen, Karuna, Lundbeck, Lyndra, Medavante-ProPhase, Merck, Neurocrine, Novartis, Noven, Otsuka, Ovid, Relmada, Reviva, Sage, Sunovion, Supernus, Teva, and University of Arizona and one-off ad hoc consulting for individuals/entities conducting marketing, commercial, or scientific scoping research; speaker for AbbVie/Allergan, Acadia, Alkermes, Angelini, Eisai, Intra-Cellular Therapies, Janssen, Lundbeck, Neurocrine, Noven, Otsuka, Sage, Sunovion, Takeda, Teva, and CME activities organized by medical education companies such as Medscape, NACCME, NEI, Vindico, and Universities and Professional Organizations/Societies; stock ownership (small number of shares of common stock) in Bristol-Myers Squibb, Eli Lilly, J & J, Merck, Pfizer purchased > 10 years ago; stock options: Reviva; and royalties from Wiley (Editor-in-Chief, International Journal of Clinical Practice, through end 2019), UpToDate (reviewer), Springer Healthcare (book), Elsevier (Topic Editor, Psychiatry, Clinical Therapeutics). Dr Preskorn reported receiving research grants from, serving as a consultant for, being on advisory boards of, and being a member of the speakers bureaus of Alkermes, BioXcel Therapeutics, Eisai, Janssen, Novartis, Otsuka, Sunovion, and Usona Institute. Dr Lauriello reported serving on the data monitoring committee for this study; serving as a member of the speakers bureau for Roche Pharmaceuticals; serving on advisory boards for Alkermes, BioXcel Therapeutics, Indivior, and Osmotica; receiving travel funding from Alkermes; and serving as a member of the data monitoring committee for Avanir. Dr Krystal was a consultant for Aptinyx, Atai Life Sciences, AstraZeneca Pharmaceuticals, Biogen Idec, Biomedisyn Corporation, Bionomics, Limited, Boehringer Ingelheim International, Cadent Therapeutics, Clexio Bioscience, COMPASS Pathways, Limited, Concert Pharmaceuticals, Epiodyne, EpiVario, Greenwich Biosciences, Heptares Therapeutics, Limited (UK), Janssen Research & Development, Jazz Pharmaceuticals, Otsuka America Pharmaceutical, Perception Neuroscience Holdings, Spring Care, Sunovion Pharmaceuticals, Takeda Industries, Taisho Pharmaceutical Co. He served on advisory boards for Biohaven Pharmaceuticals, BioXcel Therapeutics, BlackThorn Therapeutics, Cadent Therapeutics, Cerevel Therapeutics, EpiVario, Eisai, Lohocla Research Corporation, Novartis Pharmaceuticals Corporation, PsychoGenics, Tempero Bio, and Terran Biosciences. He owns stock or stock options in Biohaven Pharmaceuticals, Sage Pharmaceuticals, Spring Care, Biohaven Pharmaceuticals Medical Sciences, BlackThorn Therapeutics, EpiVario, Terran Biosciences, and Tempero Bio, and he is on the editorial board of Biological Psychiatry. Dr Kakar was a primary investigator for the sublingual dexmedetomidine clinical research program and has consulted for Neurocrine Biosciences, Alkermes, and Otsuka. Dr Finman was a paid statistical consultant of BioXcel Therapeutics. Dr De Vivo is employed by BioXcel Therapeutics and has a patent pending related to this research. Dr Yocca is employed by BioXcel Therapeutics. Dr Risinger is employed by BioXcel Therapeutics and has patents pending issued to BioXcel for sublingual dexmedetomidine as a method to treat agitation and for methods for detection and prevention of agitation. Dr Rajachandran is employed by BioXcel Therapeutics.
Funding/support: This study was funded by BioXcel Therapeutics Inc, New Haven, Connecticut.
Role of the sponsor: BioXcel Therapeutics designed the trial, developed the protocol, and, with WCG Statistics Collaborative, Washington, DC, developed the statistical analysis plan. Data collection and management were performed by Cognitive Research Corporation. The sponsor had no access to the unblinded data until the database was locked at the end of the study. BioXcel funded editorial assistance and reviewed the manuscript.
Previous presentations: Presented at the American Psychiatric Association, May 3, 2021, Virtual; American College of Emergency Physicians, October 25, 2021, Boston, MA; Psych Congress, October 30, 2021, San Antonio, TX; Neuroscience Education Institute, November 5, 2021, Colorado Springs, CO; American Society of Health Systems Pharmacists, December 5, 2021, Virtual; American Academy of Emergency Psychiatry, December 9, 2021, Las Vegas, NV.
Acknowledgments: The authors acknowledge the editorial assistance of Christopher Caiazza; Heather Robison, MMP; and Alix Bennett, PhD, in the development of this manuscript, which was supported by BioXcel Therapeutics, New Haven, CT. Lisa Weissfeld, PhD, WCG Statistics Collaborative, Washington, DC, developed the statistical analysis plan, and James Zhou, PhD, Cognitive Research Corporation, Fort Lauderdale, FL, wrote the SAS/STAT software programming; both were paid consultants.
Supplementary material: Available at Psychiatrist.com.
Clinical Points
- Acute agitation is common among individuals with schizophrenia, and rapid management is needed to reduce distress and avoid further escalation.
- To minimize the need for physical restraints, rapid, noncoercive, noninvasive, and collaborative treatment approaches are preferred.
- Self-administered sublingual dexmedetomidine reduced symptoms of agitation and was generally well tolerated.
References (27)

- Powell G, Caan W, Crowe M. What events precede violent incidents in psychiatric hospitals? Br J Psychiatry. 1994;165(1):107–112. PubMed CrossRef
- Garriga M, Pacchiarotti I, Kasper S, et al. Assessment and management of agitation in psychiatry: expert consensus. World J Biol Psychiatry. 2016;17(2):86–128. PubMed CrossRef
- Zeller SL, Citrome L. Managing agitation associated with schizophrenia and bipolar disorder in the emergency setting. West J Emerg Med. 2016;17(2):165–172. PubMed CrossRef
- San L, Marksteiner J, Zwanzger P, et al. State of acute agitation at psychiatric emergencies in Europe: the STAGE Study. Clin Pract Epidemiol Ment Health. 2016;12(1):75–86. PubMed CrossRef
- Roberts J, Gracia Canales A, Blanthorn-Hazell S, et al. Characterizing the experience of agitation in patients with bipolar disorder and schizophrenia. BMC Psychiatry. 2018;18(1):104. PubMed CrossRef
- Pompili M, Ducci G, Galluzzo A, et al. The management of psychomotor agitation associated with schizophrenia or bipolar disorder: a brief review. Int J Environ Res Public Health. 2021;18(8):4368. PubMed CrossRef
- Holloman GH Jr, Zeller SL. Overview of Project BETA: best practices in evaluation and treatment of agitation. West J Emerg Med. 2012;13(1):1–2. PubMed CrossRef
- Occupation Safety & Health Administration (OSHA). Work Place Violence in Healthcare - Understanding the Challenge. OSHA website. https://www.osha.gov/Publications/OSHA3826.pdf. Published 2015. Accessed July 26, 2021.
- Richmond JS, Berlin JS, Fishkind AB, et al. Verbal de-escalation of the agitated patient: consensus statement of the American Association for Emergency Psychiatry Project BETA De-escalation Workgroup. West J Emerg Med. 2012;13(1):17–25. PubMed CrossRef
- Dundar Y, Greenhalgh J, Richardson M, et al. Pharmacological treatment of acute agitation associated with psychotic and bipolar disorder: a systematic review and meta-analysis. Hum Psychopharmacol. 2016;31(4):268–285. PubMed CrossRef
- Wilson MP, Pepper D, Currier GW, et al. The psychopharmacology of agitation: consensus statement of the american association for emergency psychiatry project Beta psychopharmacology workgroup. West J Emerg Med. 2012;13(1):26–34. PubMed CrossRef
- Kersting XAK, Hirsch S, Steinert T. Physical harm and death in the context of coercive measures in psychiatric patients: a systematic review. Front Psychiatry. 2019;10:400. PubMed CrossRef
- Chieze M, Hurst S, Kaiser S, et al. Effects of seclusion and restraint in adult psychiatry: a systematic review. Front Psychiatry. 2019;10:491. PubMed CrossRef
- Citrome L. Comparison of intramuscular ziprasidone, olanzapine, or aripiprazole for agitation: a quantitative review of efficacy and safety. J Clin Psychiatry. 2007;68(12):1876–1885. PubMed CrossRef
- Faden J, Citrome L. Examining the safety, efficacy, and patient acceptability of inhaled loxapine for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults. Neuropsychiatr Dis Treat. 2019;15:2273–2283. PubMed CrossRef
- Igalmi (sublingual dexmedetomidine) [package insert]. New Haven, CT: BioXcel Therapeutics Inc; April 2022.
- Precedex (dexmedetomidine HCL) [package insert]. Lake Forest, IL: Hospira; Revised May 2021.
- Yocca FD, De Vivo M, Seth S, et al. Dexmedetomidine–highly favorable pharmacokinetic and pharmacological features for a CNS therapeutic drug. Neuropsychopharmacology. 2019;44(44):352.
- Preskorn SH, Risinger R, Kakar R. et al. Double-blind, placebo-controlled, single ascending dose, study to determine the efficacy, safety, and pharmacokinetics of a BXCL501 (sublingual dexmedetomidine) in agitation associated with schizophrenia or related. Neuropsychopharmacology. 2019;44:114.
- Preskorn SH, Zeller S, Citrome L, et al. Effect of sublingual dexmedetomidine vs placebo on acute agitation associated with bipolar disorder: a randomized clinical trial. JAMA. 2022;327(8):727–736. PubMed CrossRef
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Fifth Edition. Arlington, VA: American Psychiatric Association; 2013.
- Sheehan DV, Lecrubier Y, Sheehan KH, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59(suppl 20):22–33, quiz 34–57. PubMed
- Montoya A, Valladares A, Lizán L, et al. Validation of the excited component of the Positive and Negative Syndrome Scale (PANSS-EC) in a naturalistic sample of 278 patients with acute psychosis and agitation in a psychiatric emergency room. Health Qual Life Outcomes. 2011;9(1):18. PubMed CrossRef
- Busner J, Targum SD. The clinical global impressions scale: applying a research tool in clinical practice. Psychiatry (Edgmont). 2007;4(7):28–37. PubMed
- Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266–1277. PubMed CrossRef
- Patel MX, Sethi FN, Barnes TRE, et al. Joint BAP NAPICU evidence-based consensus guidelines for the clinical management of acute disturbance: de-escalation and rapid tranquillisation. J Psychiatr Intensive Care. 2018;14(2):89–132. CrossRef
- Martínez-Raga J, Amore M, Di Sciascio G, et al. 1st International Experts’ meeting on agitation: conclusions regarding the current and ideal management paradigm of agitation. Front Psychiatry. 2018;9:54. PubMed CrossRef
This PDF is free for all visitors!





