Background: This 8-week, randomized, double-blind, placebo-controlled study, conducted August 2010-May 2012 in the United States, evaluated the safety and efficacy of vortioxetine 10 mg and 15 mg in patients with major depressive disorder (MDD). The mechanism of action of vortioxetine is thought to be related to direct modulation of serotonin (5-HT) receptor activity and inhibition of the serotonin transporter.
Method: Adults aged 18-75 years with MDD (DSM-IV-TR) and Montgomery-Asberg Depression Rating Scale (MADRS) total score ≥ 26 were randomized (1:1:1) to receive vortioxetine 10 mg or 15 mg or placebo once daily, with the primary efficacy end point being change from baseline at week 8 in MADRS analyzed by mixed model for repeated measures. Adverse events were recorded during the study, suicidal ideation and behavior were assessed using the Columbia-Suicide Severity Rating Scale (C-SSRS), and sexual dysfunction was assessed using the Arizona Sexual Experience (ASEX) scale.
Results: Of the 1,111 subjects screened, 469 subjects were randomized: 160 to placebo, 157 to vortioxetine 10 mg, and 152 to vortioxetine 15 mg. Differences from placebo in the primary efficacy end point were not statistically significant for vortioxetine 10 mg or vortioxetine 15 mg. Nausea, headache, dry mouth, constipation, diarrhea, vomiting, dizziness, and flatulence were reported in ≥ 5% of subjects receiving vortioxetine. Discontinuation due to adverse events occurred in 7 subjects (4.4%) in the placebo group, 8 (5.2%) in the vortioxetine 10 mg group, and 12 (7.9%) in the vortioxetine 15 mg group. ASEX total scores were similar across groups. There were no clinically significant trends within or between treatment groups on the C-SSRS, laboratory values, electrocardiogram, or vital sign parameters.
Conclusions: In this study, vortioxetine did not differ significantly from placebo on MADRS total score after 8 weeks of treatment in MDD subjects.
Trial Registration: ClinicalTrials.gov identifier: NCT01179516
This work may not be copied, distributed, displayed, published, reproduced, transmitted, modified, posted, sold, licensed, or used for commercial purposes.
By downloading this file, you are agreeing to the publisher’s Terms & Conditions.
A Randomized, Double-Blind, Placebo-Controlled Study of the Efficacy and Safety of 2 Doses of Vortioxetine in Adults With
Major Depressive Disorder
ABSTRACT
Background: This 8-week, randomized, double-blind, placebo-controlled study, conducted August 2010–May 2012 in the United States, evaluated the safety and efficacy of vortioxetine 10 mg and 15 mg in patients with major depressive disorder (MDD). The mechanism of action of vortioxetine is thought to be related to direct modulation of serotonin (5-HT) receptor activity and inhibition of the serotonin transporter.
Method: Adults aged 18–75 years with MDD (DSM-IV-TR) and Montgomery-Asberg Depression Rating Scale (MADRS) total score ≥ 26 were randomized (1:1:1) to receive vortioxetine 10 mg or 15 mg or placebo once daily, with the primary efficacy end point being change from baseline at week 8 in MADRS analyzed by mixed model for repeated measures. Adverse events were recorded during the study, suicidal ideation and behavior were assessed using the Columbia-Suicide Severity Rating Scale (C-SSRS), and sexual dysfunction was assessed using the Arizona Sexual Experience (ASEX) scale.
Results: Of the 1,111 subjects screened, 469 subjects were randomized: 160 to placebo, 157 to vortioxetine 10 mg, and 152 to vortioxetine 15 mg. Differences from placebo in the primary efficacy end point were not statistically significant for vortioxetine 10 mg or vortioxetine 15 mg. Nausea, headache, dry mouth, constipation, diarrhea, vomiting, dizziness, and flatulence were reported in ≥ 5% of subjects receiving vortioxetine. Discontinuation due to adverse events occurred in 7 subjects (4.4%) in the placebo group, 8 (5.2%) in the vortioxetine 10 mg group, and 12 (7.9%) in the vortioxetine 15 mg group. ASEX total scores were similar across groups. There were no clinically significant trends within or between treatment groups on the C-SSRS, laboratory values, electrocardiogram, or vital sign parameters.
Conclusions: In this study, vortioxetine did not differ significantly from placebo on MADRS total score after 8 weeks of treatment in MDD subjects.
Trial Registration: ClinicalTrials.gov identifier: NCT01179516
J Clin Psychiatry 2015;76(5):583–591
© Copyright 2015 Physicians Postgraduate Press, Inc.
Submitted: June 23, 2014; accepted January 23, 2015
(doi:10.4088/JCP.14m09337).
Corresponding author: Madhukar H. Trivedi, MD, 6363 Forest Park Rd, Ste 13.354, Dallas, TX 75390-9119
(madhukar.trivedi@utsouthwestern.edu).
Over the last 3 decades, treatment effects have declined in studies of acute treatment of MDD, and these studies often show inconsistent results across trials of the same antidepressant.1,2 Even among agents indicated for MDD treatment, only 53% of trials demonstrate superiority over placebo.1 The reasons for such inconsistent results have not been fully elucidated and appear to be multifactorial. A recent meta-analysis1 found that the baseline severity of symptoms had a stronger correlation with study outcomes than did sample size, study duration, flexible versus fixed dosing, and geographic location of study sites. A similar positive correlation between symptom severity and outcomes was shown in another meta-analysis.2 In a third analysis, symptom severity was not a significant predictor of the difference between active treatment and placebo, whereas enrollment at academic sites was strongly predictive of positive treatment effects.3 This study suggested that raters in nonacademic settings may have a tendency to overestimate baseline scores that, in turn, could lead to higher placebo responses. Consistent with this observation, results of a study by Kobak et al4 showed that site-based raters were more likely to assign higher 17-item Hamilton Depression Rating Scale scores at baseline than centralized raters who screened patients remotely. Placebo response rates were also higher in the populations screened by the site-based raters. These findings suggest that factors associated with the study execution also influence study outcomes.
Vortioxetine is an investigational antidepressant currently approved for the treatment of major depressive disorder (MDD). The mechanism of action of vortioxetine is thought to be related to its multimodal activity, a combination of 2 pharmacologic modes of action: direct modulation of serotonin (5-HT) receptor activity and inhibition of the 5-HT transporter. In vitro studies indicate that vortioxetine is a 5-HT3, 5-HT7, and 5-HT1D receptor antagonist, a 5-HT1B receptor partial agonist, a 5-HT1A receptor agonist, and an inhibitor of the 5-HT transporter.5,6 The precise contribution of the individual targets to the observed pharmacodynamic profile remains unclear. However, preclinical data suggest that the targets interact in a complex fashion, leading to modulation of neurotransmission in several systems, including serotonin, norepinephrine, dopamine, histamine, and acetylcholine systems, within the rat forebrain.7,8 The vortioxetine clinical development program for MDD included 5 short-term placebo-controlled studies in adults9–13 and 1 in elderly patients14 that evaluated vortioxetine doses of 1, 2.5, 5, and 10 mg. Three were positive,9,11,14 and 1 failed on the primary analysis but showed a separation from placebo on a sensitivity analysis.10 One short-term study evaluating 5 mg and a second evaluating 2.5 mg and 5 mg failed.12,13 The current study is part of a revised development program assessing vortioxetine doses up to and including 20 mg.
The primary objective of this study was to investigate the efficacy and safety of vortioxetine 10 mg/d and 15 mg/d doses versus placebo in the treatment of MDD. A remote, centralized rater system was incorporated into the study protocol to minimize the potential influence of rater bias.
METHOD
Study Design
This 8-week, randomized, double-blind, placebo-controlled, parallel-group, phase 3 study of vortioxetine 10 mg and 15 mg in subjects with MDD was conducted at 65 sites in the United States (ClinicalTrials.gov identifier: NCT01179516). Eligible subjects were randomized (1:1:1) to receive placebo, vortioxetine 10 mg, or vortioxetine 15 mg once daily during the 8-week double-blind treatment period using an interactive voice-response system (Perceptive Informatics). Subjects assigned to receive vortioxetine 15 mg received a 10-mg dose for the first week of the 8-week study. Study medication was encapsulated in Swedish-orange capsules; identical capsules containing a lactose monohydrate/magnesium stearate filler were used for placebo.
The protocol was approved by individual institutional review boards and conducted in compliance with US Food and Drug Administration code of Federal Regulations Part 21, the International Conference on Harmonization Tripartite Guidelines for Good Clinical Practice, and the World Medical Association Declaration of Helsinki. After providing signed informed consent, subjects entered a screening period for up to 14 days and, if eligible, were randomized. Enrollment began in August 2010, and the study ended on May 11, 2012. Subjects who completed the 8-week treatment period were offered the option to participate in a long-term, open-label, safety extension study if they met the selection criteria. Subjects who did not enroll in the open-label study were followed for safety for an additional 4 weeks after the last dose of study medication.
Rater Selection and Training
The centralized raters were selected based on their rating experience and underwent additional training specifically for this study. Periodically during the study, the interviews conducted by centralized raters were reviewed by standard raters to ensure interrater reliability.
Stringent criteria were used in selection of site-based raters who were experienced in administering the Hamilton Anxiety Rating Scale (HARS),15 Clinical Global Impressions-Severity of Illness scale (CGI-S),16 and Clinical Global Impressions-Improvement scale (CGI-I).16 Site-based raters’ interviewing skills were evaluated either in person at the investigator meeting or via videotaped interview. Performance on rating a standard interview and interviewing skills were considered when certifying raters as eligible to rate subjects.
Subjects
Adult men and nonpregnant women (aged 18 to 75 years, inclusive) were eligible for participation in the study if they had a primary diagnosis of recurrent MDD as defined by DSM-IV-TR code 296.3x,17 a reported duration of current major depressive episode ≥ 3 months, a Montgomery-Asberg Depression Rating Scale (MADRS)18 total score ≥ 26 at screening and baseline, and a CGIS total score ≥ 4 at screening and baseline. Subjects were excluded from study participation if they had received any investigational compound < 30 days before screening or 5 half-lives prior to screening; had received vortioxetine in a previous clinical study; had depressive symptoms considered resistant to 2 or more adequate antidepressant treatments; had any concurrent psychiatric disorder other than MDD or prior history of psychiatric disorders such as manic or hypomanic episode, schizophrenia, or substance abuse disorder; had a significant suicide risk in the opinion of the investigator or a score of ≥ 5 on item 10 of the MADRS; or had a history of neurologic disorders or medically unstable conditions (eg, renal or hepatic impairment, cardiovascular, pulmonary, or gastrointestinal disorders). All subjects were required to have a 2-week (or longer depending on drug half-life) washout period for any psychoactive medications prior to screening.
Assessments
Subjects were screened ≤ 14 days prior to randomization. The initial screening included site-based assessments to identify those subjects who met all inclusion criteria and did not meet any of the exclusion criteria. Each subject’s eligibility was confirmed by the Structured Clinical Interview for DSM Disorders–Clinical Trial version,17 and the MADRS18 was administered remotely by centralized raters utilizing videoconferencing technology. The technology linked the subject with the centralized rater over an Internet protocol (IP) virtual private network. There was no connection or access to or from any other device, including the Internet. To establish a secure and encrypted connection, the centralized rater selected the remote site from a study directory of preconfigured IP numbers. This system permitted the centralized rater to perform all aspects of the assessments. In addition to the screening visit, centralized raters remotely conducted MADRS assessments at all subsequent study visits.
The centralized raters were blinded to the study design (primary end point, duration, number of treatment groups, drug involved, dose, other scales utilized), inclusion/exclusion criteria, and visit number. In addition, subjects were assessed by different centralized raters during treatment to minimize the possibility of developing a therapeutic relationship or the anticipation or expectation of change due to study participation and to allow for blinding of visit number.
The MADRS and CGI-S scores were measured at screening, baseline, and weeks 1, 2, 4, 6, and 8. HARS scores were examined at baseline and weeks 1, 2, 4, 6, and 8, and CGI-I scores, at weeks 1, 2, 4, 6, and 8. Sheehan Disability Scale (SDS)19 score was determined at baseline and at weeks 6 and 8. The Cognitive and Physical Functioning Questionnaire (CPFQ)20 and Work Productivity and Activity Impairment inventory (WPAI)21 were conducted at baseline and week 8.
Safety assessments were performed at screening and at the visits listed as follows. Adverse events (AEs) were assessed at baseline and weeks 1, 2, 4, 6, and 8, and follow-up and were coded by system organ class and preferred term using the Medical Dictionary for Regulatory Activities, Version 11.1. Vital signs were measured at baseline and weeks 1, 2, 4, 6, and 8, and weight was measured at baseline and weeks 4 and 8. Electrocardiograms (ECGs) and laboratory tests were measured at baseline and weeks 4 and 8, and physical examinations were performed at screening and week 8. Suicidal ideation and behavior were prospectively monitored using the Columbia-Suicide Severity Rating Scale (C-SSRS),22 which was administered at screening, baseline, and weeks 1, 2, 4, 6, and 8. The Arizona Sexual Experience Scale (ASEX),23 a validated 5-item scale with versions specific to gender and sexual orientation, was used to assess the impact of treatment on sexual function. The ASEX scale was completed at baseline and weeks 1, 2, 4, 6, and 8.
Statistical Analysis
The safety set included all subjects who received at least 1 dose of study medication; the full analysis set (FAS) comprised all randomized subjects who received at least 1 dose of study drug and had at least 1 postbaseline value for the primary efficacy assessment. Data analysis and descriptive and inferential statistics tabulations were performed using SAS System, Version 9.1.3, on a UNIX platform.
The primary efficacy end point was the change from baseline in MADRS total score at week 8. Comparisons between placebo and the vortioxetine 10 mg and 15 mg treatment groups were performed using mixed model for repeated measures (MMRM) analysis of covariance (ANCOVA), with treatment, center, week, treatment-by-week interaction, and baseline MADRS total score by week as fixed effects. The effect of each treatment was allowed to vary freely, and an unstructured covariance model was assumed. Based on a missing-at-random assumption, this analysis was performed using observed case (OC) data. In a sensitivity analysis, the change from baseline in MADRS total score after 8 weeks of treatment was analyzed using ANCOVA, with treatment and center as fixed factors and baseline MADRS total score as covariate, and using the last-observation-carried forward (LOCF) and OC methods.
MADRS response (≥ 50% decrease in the MADRS total score from baseline), MADRS remission (MADRS total score ≤ 10), CGI-S remission (CGI-S score ≤ 2), and CGI-I response (CGI-I score ≤ 2) were analyzed at all time points by logistic regression adjusting for baseline score and treatment by both LOCF and OC methods. Changes from baseline in the HARS, CGI-I, and CGI-S scores were analyzed by study visit using both MMRM and ANCOVA, similar to the method used for the primary variable MADRS.
To control for the 2-sided type I errors over all key end points, a prespecified sequential testing procedure was applied over all key end points to compare vortioxetine 10 mg and 15 mg doses with placebo. The efficacy end points were tested in the following sequence at a Bonferroni-corrected significance level of .025. As soon as an end point was nonsignificant at .025, formal statistical testing was stopped for all subsequent end points for that dose, and all P values < .05 for that dose were considered nominal and described as separated from placebo.
- Primary end point: change from baseline in MADRS total score at week 8 (MMRM)
- MADRS response (≥ 50% decrease in the MADRS total score from baseline) rate at week 8 (LOCF)
- Mean CGI-I score at week 8 (MMRM)
- Change from baseline in MADRS total score at week 8 in subjects with baseline HARS score ≥ 20 (MMRM)
- MADRS remission (MADRS total score ≤ 10) rate at week 8 (LOCF)
- Change from baseline in SDS total score at week 8 (MMRM)
Change from baseline in MADRS total score was assessed in subjects with moderate depression (baseline MADRS total scores ≤ 34) and those with severe depression (baseline MADRS total scores > 34) in a prespecified subgroup analysis.
Descriptive statistics were reported for AEs, vital signs, weight, ECG findings, laboratory values, and physical examination findings.
The C-SSRS was summarized using descriptive statistics by study visit, number of subjects with positive reports at baseline and during treatment, and a shift-table to demonstrate changes in C-SSRS scores from baseline during treatment.
The main analysis of the ASEX was to assess the number of subjects who were without sexual dysfunction at baseline and developed it any time during the study period. Sexual dysfunction was defined as an ASEX total score of ≥ 19, a score ≥ 5 on any item, or a score ≥ 4 on any 3 items.23
The vortioxetine treatment groups were compared with placebo. Confidence intervals (95% 2-sided) for the differences between the incidence rates for each treatment group and placebo were constructed using the normal approximation to the binomial (percent treatment group minus percent placebo).
RESULTS
Subjects
As shown in Figure 1, of 1,111 subjects screened, 469 were randomized: 160 to placebo, 157 to vortioxetine 10 mg, and 152 to vortioxetine 15 mg. More than half of subjects screened were not randomized, largely due to not meeting inclusion criteria (19.5%) or meeting exclusion criteria (36.2%). The ranges of baseline scores were relatively broad in all 3 treatment groups as reflected in the standard deviation.
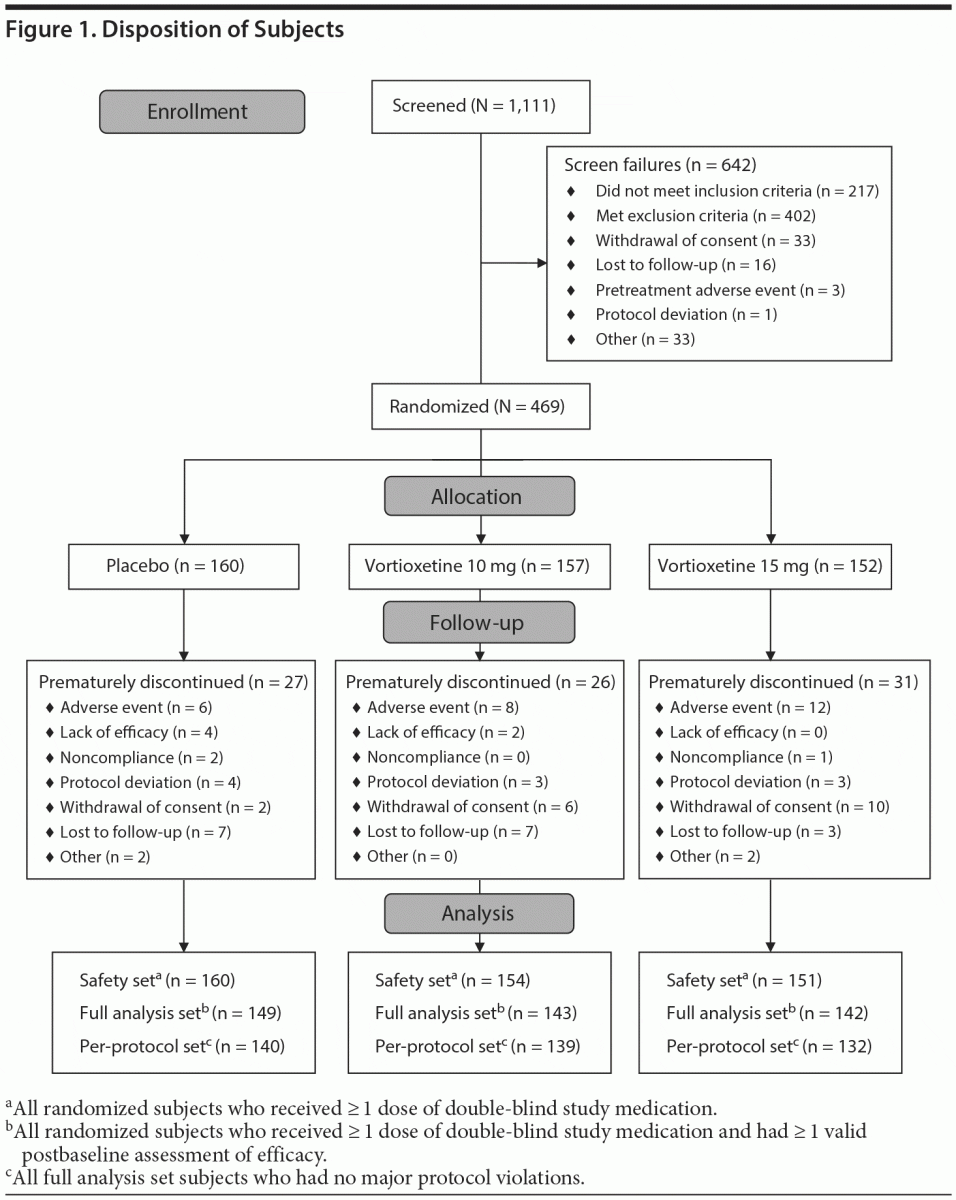
Demographics and baseline clinical characteristics were balanced across treatment groups in most categories (Table 1). Approximately 30% of the overall population was male, and the mean age was 45.1 years. The mean body mass index was 31.07 kg/m2. The mean (± SD) duration of the current depressive episode was 43.2 (± 42.5) weeks in the placebo group, 39.9 (± 32.7) weeks in the vortioxetine 10 mg group, and 46.2 (± 43.4) weeks in the vortioxetine 15 mg group. Most subjects (64.8%) had experienced 1 to 3 previous major depressive episodes at study entry. Overall, 90.4% of subjects received pharmacotherapy for MDD at some time prior to study entry. More individuals (13.2%) in the vortioxetine 15 mg group did not receive pharmacotherapy compared with the placebo (6.9%) and vortioxetine 10 mg (5.7%) groups.
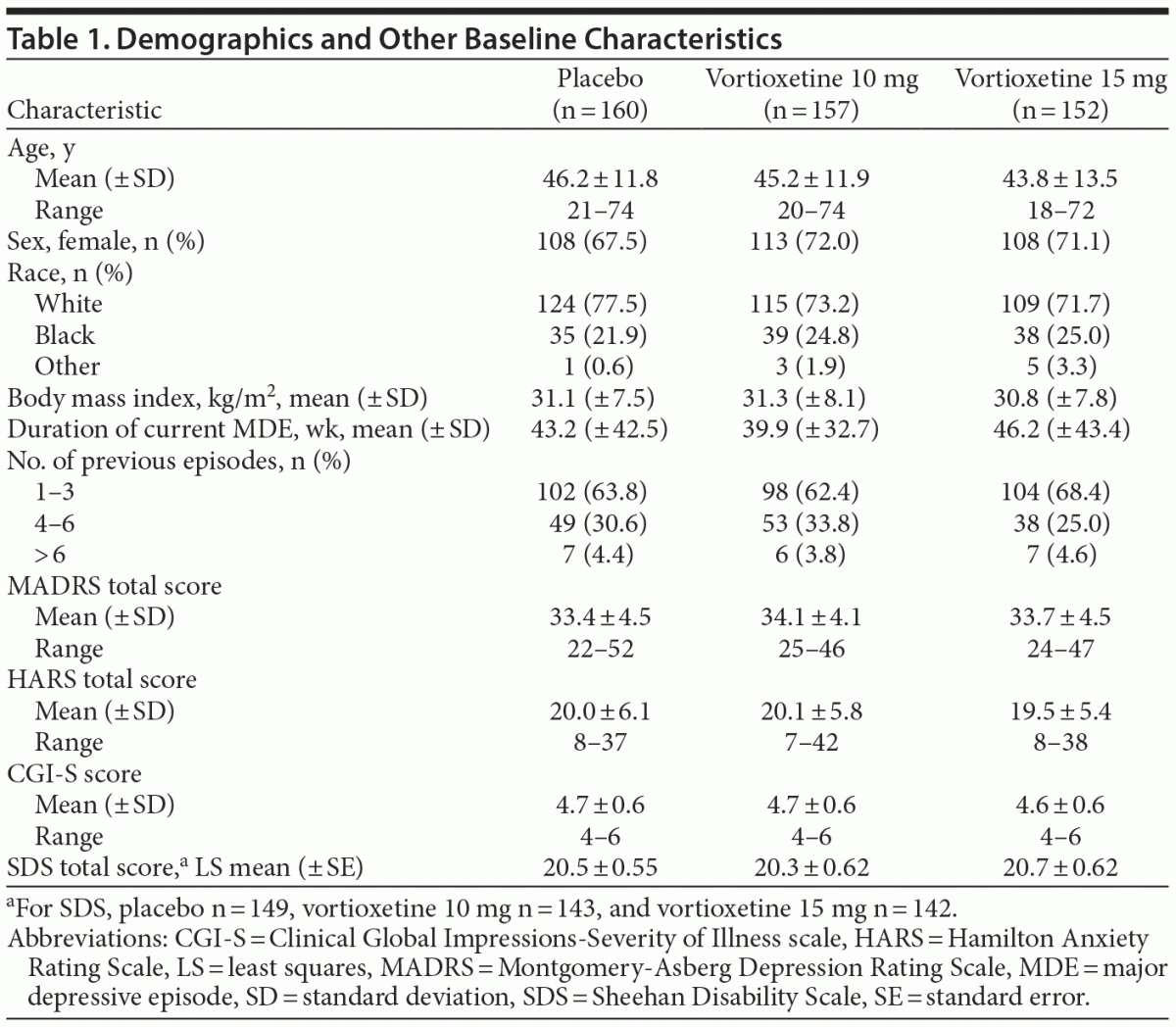
Based on pill counts (number of pills dispensed minus number returned), compliance rates were > 99% in all treatment groups.
Primary Efficacy End Point
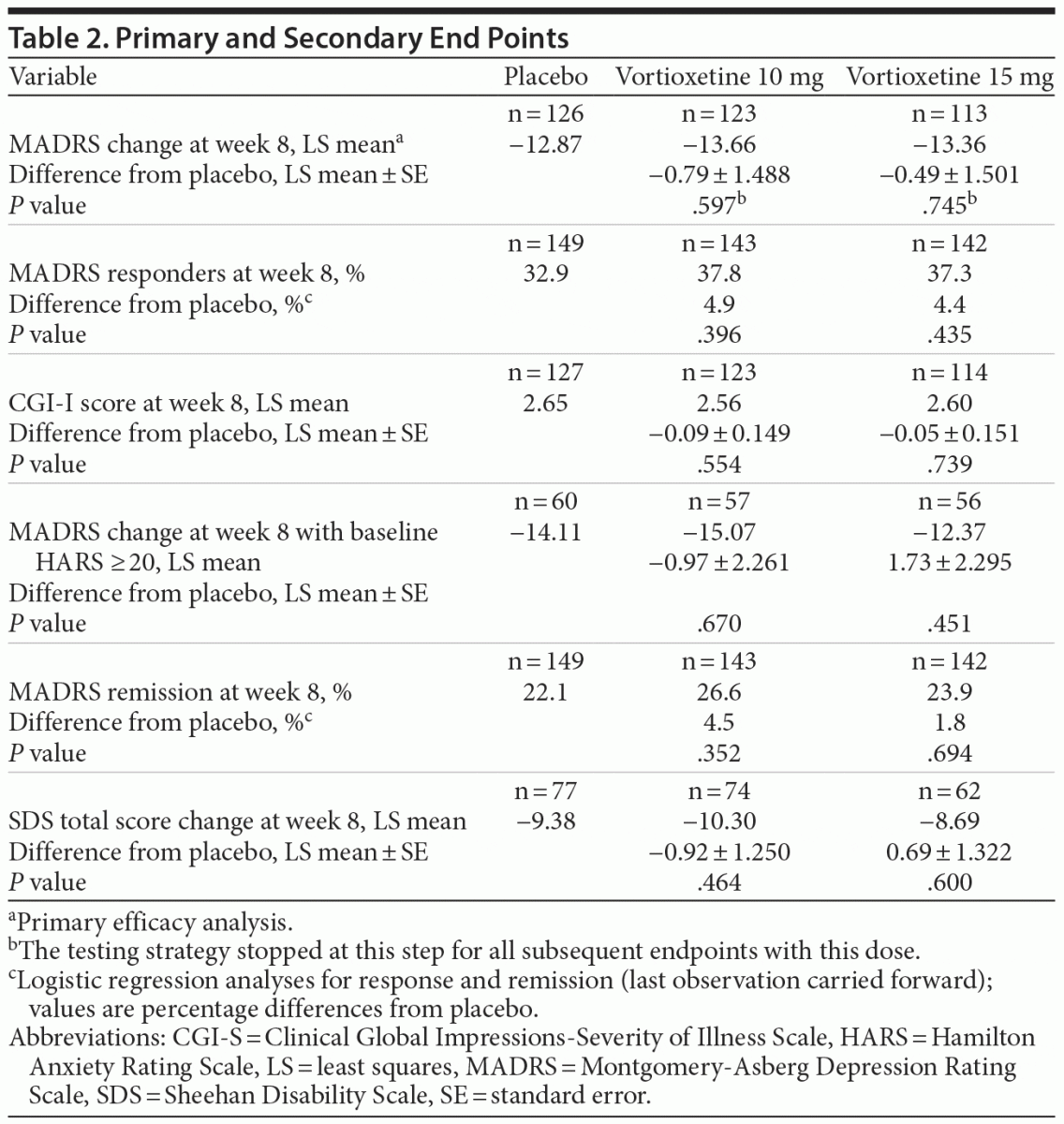
Differences from placebo in mean change from baseline MADRS scores were not statistically significant for the vortioxetine 10 mg group (difference from placebo, −0.79) or the vortioxetine 15 mg group (difference from placebo, −0.49) using the MMRM analysis (Table 2; Figure 2). Analysis using ANCOVA LOCF or OC did not alter the significance of the result.
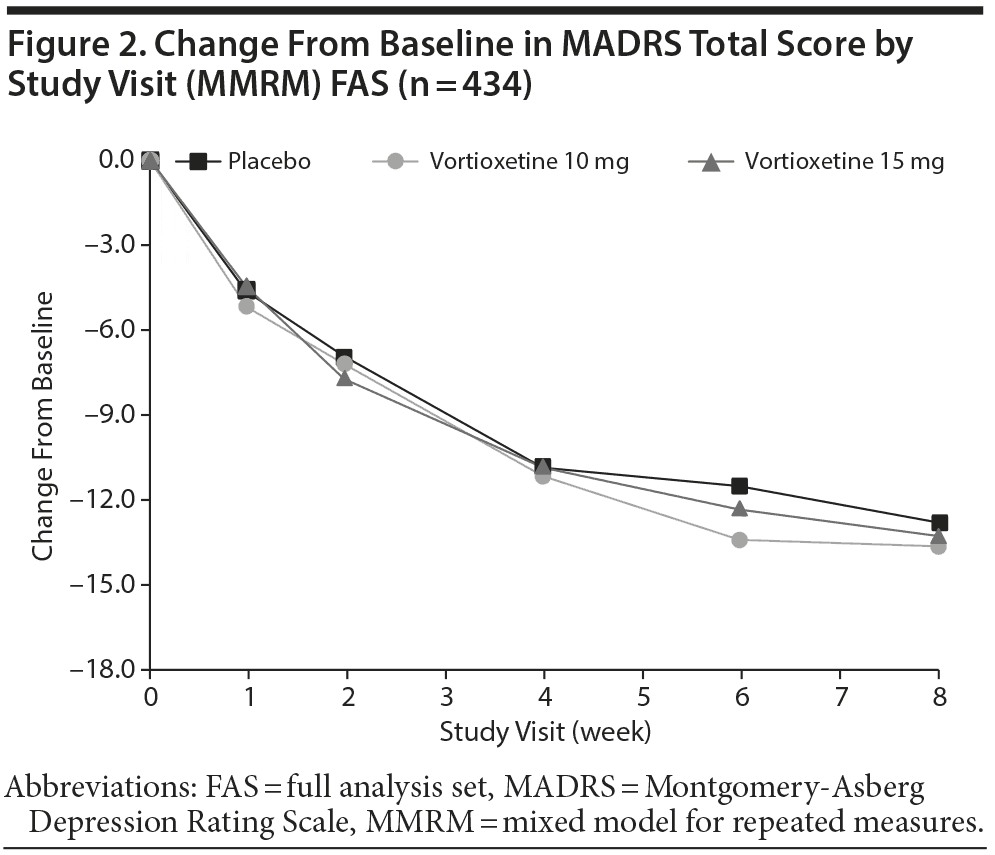
Secondary Efficacy Outcomes
For all 5 key secondary efficacy end points (Table 2), the results were similar between the 2 vortioxetine (10 mg and 15 mg) groups, and differences from placebo did not reach statistical significance at the .025 level. Outcomes were numerically superior to placebo in the vortioxetine groups for most key secondary end points.
There were no statistically significant differences between placebo and either of the vortioxetine treatment groups for either functional improvement measure assessed. The improvements in the SDS total scores at week 8 with vortioxetine 10 mg were numerically greater than with placebo or vortioxetine 15 mg; however, neither vortioxetine dose separated significantly from placebo. A similar pattern was observed with change from baseline in WPAI subscale scores (data not shown). Vortioxetine 10 mg showed numerically greater improvements compared with placebo in all WPAI subscales; however, neither vortioxetine dose separated significantly from placebo. Both vortioxetine doses showed numerically greater improvement in CPFQ scores compared with placebo. Neither difference was statistically significant (data not shown).
Subgroup Analysis
When the changes from baseline in MADRS total scores were analyzed in severely depressed subjects (baseline MADRS score > 34, n = 194), vortioxetine 15 mg separated from placebo (−17.9 [n = 54] vs −12.3 [n = 45], nominal P = .034), whereas vortioxetine 10 mg did not (−15.5 [n = 66], nominal P = .20). In contrast, neither vortioxetine treatment group separated from placebo in subjects with baseline MADRS total scores ≤ 34 (n = 240).
Safety Variables
Nausea, dry mouth, constipation, diarrhea, vomiting, and flatulence were reported in ≥ 5% of subjects in either vortioxetine treatment group and at a greater rate than in the placebo group (Table 3). No trends or clinically meaningful changes in ECG, vital sign, or clinical laboratory test data occurred during the study.
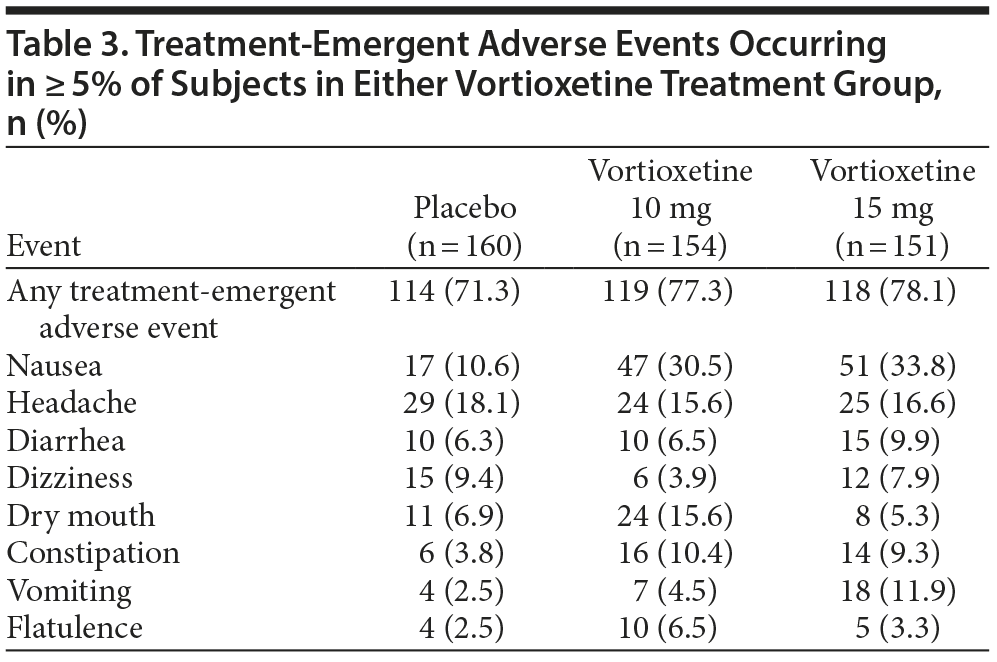
Discontinuation due to AEs occurred for 7 (4.4%) subjects in the placebo group, 8 (5.2%) in the vortioxetine 10 mg group, and 12 (7.9%) in the vortioxetine 15 mg group. Two serious AEs that led to premature withdrawal were reported during the study: lumbar radiculopathy (in the placebo group) and suicidal ideation (in the 10 mg group); both events were considered unrelated to study treatment. There were no deaths.
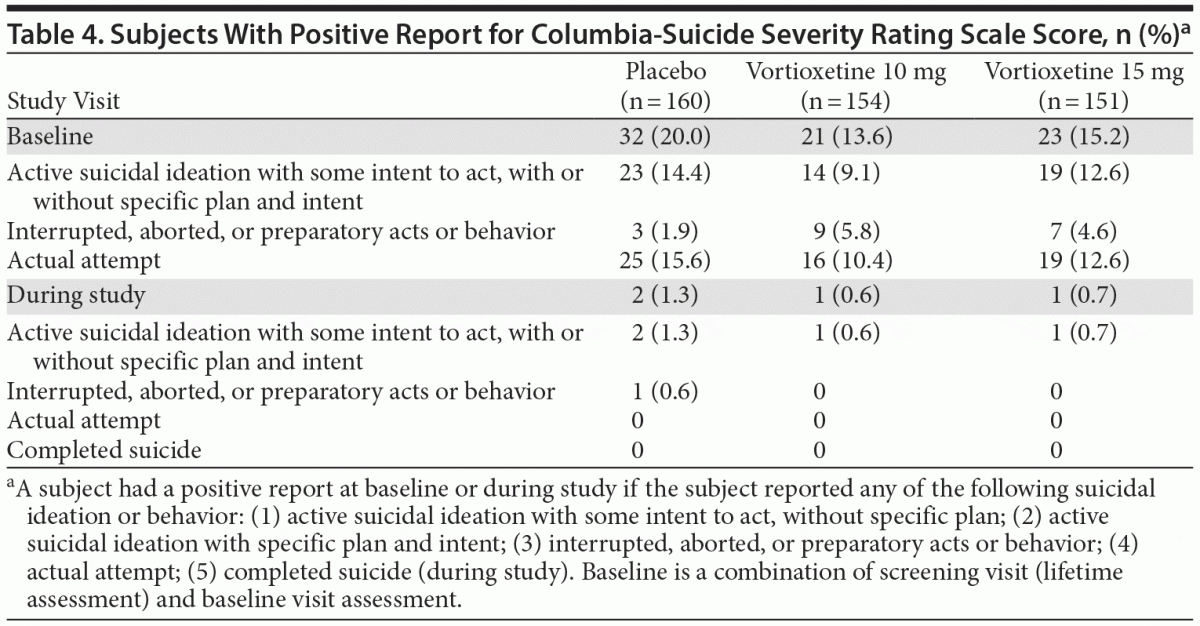
At the baseline lifetime assessment using the C-SSRS, approximately 9%–14% of subjects across all treatment groups reported active suicidal ideation; approximately 2%–6% had some type of preparatory behavior, including interrupted or aborted attempts; and between 10% and 16% had made an actual suicide attempt. As shown in Table 4, during the study, 2 placebo subjects and 1 subject in each vortioxetine group had suicidal ideation with intent to act, and 1 placebo subject reported a preparatory act (the subject bought unspecified pills to overdose but never carried out the intent). Only 1 subject withdrew from the study; the others remained in the study after evaluation by the investigator, and the blind was unbroken. No subject attempted suicide during the study.
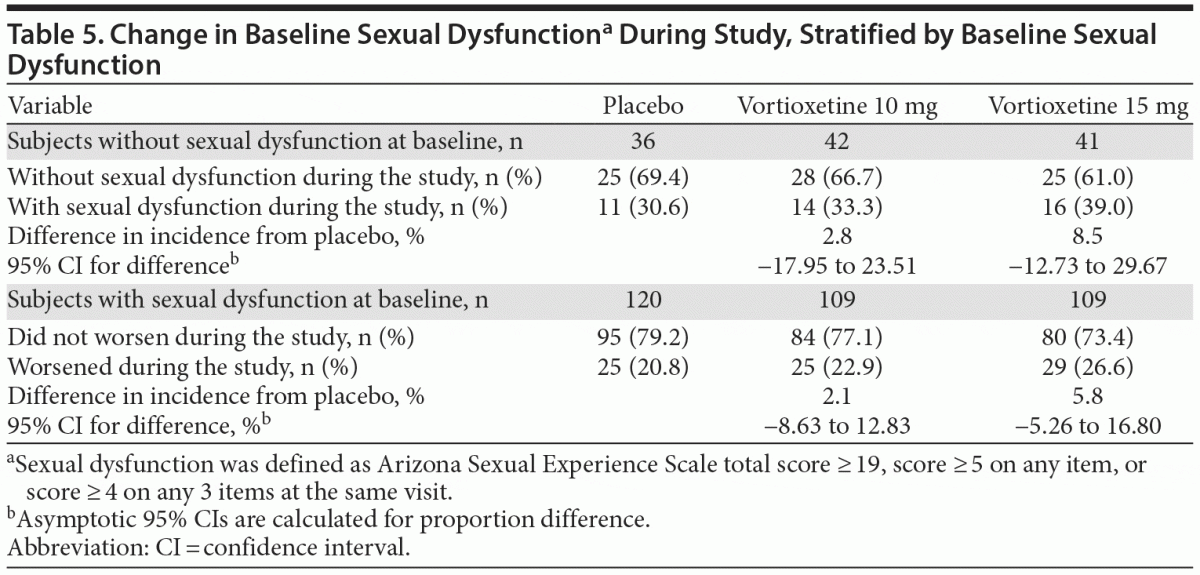
At baseline, ASEX scores were normal for 36 (23%) in the placebo group, 42 (27%) in the vortioxetine 10 mg group, and 41 (27%) in the vortioxetine 15 mg group (Table 5). Among the group with normal function at baseline, 11 (30.6%), 14 (33.3%), and 16 (39.0%) experienced sexual dysfunction at some time during the study in the placebo, vortioxetine 10 mg, and vortioxetine 15 mg groups, respectively. Differences from placebo were not statistically different for either vortioxetine dose.
DISCUSSION
These results contrast with 3 earlier studies showing significant benefit with vortioxetine 5 mg and 10 mg doses.9,11,14 In this study, the change from baseline in MADRS total score was not statistically significantly superior to placebo with vortioxetine 10 mg or 15 mg. The placebo response for the primary efficacy end point was relatively high in this trial (−12.87) and may have contributed to the lack of statistical significance. Although results for most key secondary end points, as well as functional measures (SDS, WPAI, and CPFQ scores), showed numerically better outcomes with vortioxetine, none was statistically significantly superior to placebo. The study did not include an active reference arm, so it is not possible to determine whether the trial itself failed.
Two meta-analyses1,2 of clinical trials in MDD determined that the probability of achieving a significant separation from placebo increased with severity of baseline symptoms. However, symptom severity is an unlikely explanation for nonsignificant outcomes in the current study. Subjects were required to be moderately to severely depressed (baseline MADRS total score ≥ 26), and the mean baseline MADRS total scores were 3 MADRS points higher than in a previously published study showing efficacy at the 5 mg and 10 mg doses.11 However, a prespecified subgroup analysis reported here showed a significant reduction in baseline MADRS total score in severely depressed individuals (MADRS > 34) receiving vortioxetine 15 mg compared with those receiving placebo.
In a third analysis,3 symptom severity was not a significant predictor of the difference between active treatment and placebo, whereas enrollment at academic sites was strongly predictive of positive treatment effects. It has been hypothesized that reliance on enrollment at nonacademic sites may lead to inflated depression scores at baseline (prior to randomization), which in turn may contribute to higher placebo response rates.4 A study in which the same subject group was screened by site-based and centralized raters found that the site-based raters were more likely to assign higher 17-item Hamilton Depression Rating Scale scores at screening.4 The mean change from baseline for the placebo group as assessed by site-based raters was significantly higher (7.52) versus change assessed by the centralized raters (3.18; P < .001). In addition, a meta-analysis24 of pediatric MDD trials found that clinicians’ anticipation that subjects will meet the inclusion criteria can strongly influence characteristics of the study population.
To improve assessment accuracy from screening through the study end, this trial protocol was streamlined to reduce scale assessment burden on site as well as subjects. More stringent criteria were used in selection and certification of site raters, including evaluation of their interview skills. The baseline values and primary end point (MADRS) were measured by a centralized rater. The use of centralized rating in this trial did not improve signal detection and did not seem to address the underlying issue of failed trials in mood disorders.
To compensate for the slower recruitment rate due to greater number of screen failures, the original study protocol plan for 35 study sites was doubled to include 70 sites overall, and 65 sites actually enrolled subjects. The target of 150 subjects was met for all 3 treatment groups. The sample size calculation used for the current trial was based on an anticipated treatment effect of a 3.5-point decline in MADRS total score, which is less than the effect size observed in the previous positive trials.9,11,14
These efforts appear to have increased screen failure rates. Only 42% of the subjects who were screened for this study were eligible for participation; rates for failure to meet inclusion criteria and for meeting exclusion criteria both increased. This is a substantially smaller percentage of eligible screened subjects than reported in earlier published vortioxetine studies (range, 71%–84%),11,12,14 most likely due to the use of centralized raters rather than screening of a vastly different population. In the earlier trials, 12%–21% of subjects met the exclusion criteria and < 5% did not meet inclusion criteria, compared with 36.2% and 19.3% deemed ineligible here on the basis of exclusion and inclusion criteria, respectively.
It is interesting to note that the standard deviations for mean baseline MADRS scores were larger than in other vortioxetine trials. This finding was unexpected. One possible explanation for this difference is that improved rater training resulted in more nuanced and thus diverse scores. Also, it is important to consider that the centralized raters were more stringent, but not more accurate, than the site raters in their assessments. Despite the additional measures to improve trial conduct, these results demonstrate that signal detection in MDD trials is difficult and no single methodology can guarantee trial success, even for drugs that have demonstrated efficacy in previous trials.
The current study contributes to the safety and tolerability profile of vortioxetine. Although vortioxetine was generally safe and well tolerated, the incidence of nausea was higher in this study than in others. Nausea was transient and generally mild to moderate in intensity. The AE discontinuation rates were similar between the placebo and vortioxetine 10 mg groups and higher in the vortioxetine 15 mg group. There were no serious AEs attributed to study drug, and no deaths occurred in any treatment group.
Suicidal ideation and behavior were prospectively monitored in this trial using the C-SSRS; 2 subjects reported such events in the placebo group, and 1 subject in each of the vortioxetine treatment groups reported events.
The change from baseline ASEX total scores did not differ from placebo with vortioxetine treatment (change from baseline scores: placebo, 1.06; vortioxetine 10 mg, −0.4; vortioxetine 15 mg, −1.33). Moreover, only 119 (26%) subjects did not have sexual dysfunction at baseline, and, within this subgroup, the incidence of treatment-emergent sexual dysfunction did not appear to differ across treatment groups. However, it is important to note that the sample number is too small to draw any conclusions. In both men and women, ASEX scores were similar in the placebo and vortioxetine groups at study end. In the overall population and in each of the subgroups analyzed, there was a numerically greater improvement in baseline ASEX scores with vortioxetine 15 mg compared with vortioxetine 10 mg. Whether this observation is limited to this data set, is a reflection of some underlying mechanism, or is related to dosing remains to be determined.
CONCLUSIONS
In contrast with findings of other trials, 3 of which evaluated the 10-mg dose, vortioxetine did not demonstrate a significant difference from placebo in symptomatic or functional improvement in this phase 3 study despite efforts to maximize signal detection. As in other antidepressant trials, the reasons for the failed results are difficult to ascertain. However, the safety and tolerability results provide further evidence that vortioxetine has a favorable safety and tolerability profile.
Drug names: vortioxetine (Brintellix).
Author affiliations: CNS Medicine (Dr Mahableshwarkar), Clinical Science (Ms Jacobsen), Pharmacovigilance (Dr Serenko), and CNS Statistics (Dr Chen), Takeda Development Center Americas, Deerfield, Illinois; and Division of Mood Disorders, University of Texas Southwest Medical Center, Dallas (Dr Trivedi).
Author contributions: Drs Mahableshwarkar, Serenko, Chen, and Trivedi and Ms Jacobsen each contributed to the concept, data analysis, drafting, critical revisions, and approval of the article.
Potential conflicts of interest: Dr Mahableshwarkar is an employee of Takeda Development Center Americas and holds stock in GlaxoSmithKline, Johnson & Johnson, and Pfizer. Ms Jacobsen and
Dr Chen are employees of Takeda Development Center Americas.
Dr Serenko was an employee of Takeda Development Center Americas at the time the study was conducted. Dr Trivedi has received consulting fees from Abbott, Abdi Ibrahim, Akzo, Alkermes, AstraZeneca, Axon Advisors, Bristol-Myers Squibb, Cephalon, CME Institute of Physicians Postgraduate Press, Eli Lilly, Evotek, Fabre-Kramer, Forest, GlaxoSmithKline, Janssen, Johnson & Johnson, Libby, Lundbeck, Meade Johnson, MedAvante, Medtronic, Neuronetics, Otsuka, Pamlab, Parke-Davis, Pfizer, PGxHealth, Rexahn, Sepracor, Shire, Sierra, Takeda, Tai Medical/Puretech Venture, Transcept, VantagePoint, and Wyeth-Ayerst and has received research support from the Agency for Healthcare Research and Quality, Corcept Therapeutics, Cyberonics, Inc, Merck, National Alliance for Research on Schizophrenia and Depression, National Institute of Mental Health, National Institute on Drug Abuse, Naurex, Novartis, Pharmacia & Upjohn, Predix (Epix), Solvay, Targacept, and Valiant.
Funding/support: This study was supported by the Takeda Pharmaceutical Company, Ltd and H. Lundbeck A/S.
Role of the sponsor: Takeda was involved in the design, investigator selection, conduct of the trial, collection of data, analysis and interpretation, and writing of the final study report. The authors had full control of the content of the manuscript.
Acknowledgments: Assistance with writing and manuscript preparation was provided by Ann C. Sherwood, PhD, of The Medicine Group
and funded by the Takeda Pharmaceutical Company, Ltd, and
H. Lundbeck A/S.
REFERENCES
1. Khin NA, Chen YF, Yang Y, et al. Exploratory analyses of efficacy data from major depressive disorder trials submitted to the US Food and Drug Administration in support of new drug applications. J Clin Psychiatry. 2011;72(4):464–472. doi:10.4088/JCP.10m06191 PubMed
2. Khan A, Bhat A, Kolts R, et al. Why has the antidepressant-placebo difference in antidepressant clinical trials diminished over the past three decades? CNS Neurosci Ther. 2010;16(4):217–226. doi:10.1111/j.1755-5949.2010.00151.x PubMed
3. Dunlop BW, Thase ME, Wun CC, et al. A meta-analysis of factors impacting detection of antidepressant efficacy in clinical trials: the importance of academic sites. Neuropsychopharmacology. 2012;37(13):2830–2836. doi:10.1038/npp.2012.153 PubMed
4. Kobak KA, Leuchter A, DeBrota D, et al. Site versus centralized raters in a clinical depression trial: impact on patient selection and placebo response. J Clin Psychopharmacol. 2010;30(2):193–197. doi:10.1097/JCP.0b013e3181d20912 PubMed
5. Westrich L, Pehrson A, Zhong H, et al. In vitro and in vivo effects for the multimodal antidepressant vortioxetine (Lu AA21004) at human and rat targets. Int J Psychiatry Clin Pract. 2012;16(suppl 1):47.
6. Bang-Andersen B, Ruhland T, Jørgensen M, et al. Discovery of 1-[2-(2,4-dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): a novel multimodal compound for the treatment of major depressive disorder. J Med Chem. 2011;54(9):3206–3221. doi:10.1021/jm101459g PubMed
7. Mørk A, Pehrson A, Brennum LT, et al. Pharmacological effects of Lu AA21004: a novel multimodal compound for the treatment of major depressive disorder. J Pharmacol Exp Ther. 2012;340(3):666–675. doi:10.1124/jpet.111.189068 PubMed
8. Pehrson AL, Cremers T, Bétry C, et al. Lu AA21004, a novel multimodal antidepressant, produces regionally selective increases of multiple neurotransmitters—a rat microdialysis and electrophysiology study. Eur Neuropsychopharmacol. 2013;23(2):133–145. doi:10.1016/j.euroneuro.2012.04.006 PubMed
9. Alvarez E, Perez V, Dragheim M, et al. A double-blind, randomized, placebo-controlled, active reference study of Lu AA21004 in patients with major depressive disorder. Int J Neuropsychopharmacol. 2012;15(5):589–600. doi:10.1017/S1461145711001027 PubMed
10. Baldwin DS, Hansen T, Florea I. Vortioxetine (Lu AA21004) in the long-term open-label treatment of major depressive disorder. Curr Med Res Opin. 2012;28(10):1717–1724. doi:10.1185/03007995.2012.725035 PubMed
11. Henigsberg N, Mahableshwarkar AR, Jacobsen P, et al. A randomized, double-blind, placebo-controlled 8-week trial of the efficacy and tolerability of multiple doses of Lu AA21004 in adults with major depressive disorder. J Clin Psychiatry. 2012;73(7):953–959. doi:10.4088/JCP.11m07470 PubMed
12. Jain R, Mahableshwarkar AR, Jacobsen PL, et al. A randomized, double-blind, placebo-controlled 6-wk trial of the efficacy and tolerability of 5 mg vortioxetine in adults with major depressive disorder. Int J Neuropsychopharmacol. 2013;16(2):313–321. doi:10.1017/S1461145712000727 PubMed
13. Mahableshwarkar AR, Jacobsen PL, Chen Y. A randomized, double-blind trial of 2.5 mg and 5 mg vortioxetine (Lu AA21004) versus placebo for
8 weeks in adults with major depressive disorder. Curr Med Res Opin. 2013;29(3):217–226. doi:10.1185/03007995.2012.761600 PubMed
14. Katona C, Hansen T, Olsen CK. A randomized, double-blind, placebo-controlled, duloxetine-referenced, fixed-dose study comparing the efficacy and safety of Lu AA21004 in elderly patients with major depressive disorder. Int Clin Psychopharmacol. 2012;27(4):215–223. doi:10.1097/YIC.0b013e3283542457 PubMed
15. Hamilton M. The assessment of anxiety states by rating. Br J Med Psychol. 1959;32(1):50–55. doi:10.1111/j.2044-8341.1959.tb00467.x PubMed
16. Guy W. Clinical Global Impressions (028-CGI). ECDEU Assessment Manual for Psychopharmacology. Revised. Rockville, MD: US Dept Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration; 1976.
17. American Psychiatric Association. Mood Disorders. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision. Washington, DC: American Psychiatric Association; 2000:369–382.
18. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134(4):382–389. doi:10.1192/bjp.134.4.382 PubMed
19. Sheehan DV, Harnett-Sheehan K, Raj BA. The measurement of disability. Int Clin Psychopharmacol. 1996;11(suppl 3):89–95. doi:10.1097/00004850-199606003-00015 PubMed
20. Fava M, Iosifescu DV, Pedrelli P, et al. Reliability and validity of the Massachusetts General Hospital Cognitive and Physical Functioning Questionnaire. Psychother Psychosom. 2009;78(2):91–97. doi:10.1159/000201934 PubMed
21. Reilly MC, Zbrozek AS, Dukes EM. The validity and reproducibility of a work productivity and activity impairment instrument. Pharmacoeconomics. 1993;4(5):353–365. doi:10.2165/00019053-199304050-00006 PubMed
22. Posner K, Brent D, Lucas C, et al. Columbia-Suicide Severity Rating Scale (C-SSRS). New York, NY: Columbia University Medical Center; 2008.doi:10.1176/ajp.2007.164.7.1035 PubMed
23. McGahuey CA, Gelenberg AJ, Laukes CA, et al. The Arizona Sexual Experience Scale (ASEX): reliability and validity. J Sex Marital Ther. 2000;26(1):25–40. doi:10.1080/009262300278623 PubMed
24. Mundt JC, Greist JH, Jefferson JW, et al. Is it easier to find what you are looking for if you think you know what it looks like? J Clin Psychopharmacol. 2007;27(2):121–125. doi:10.1097/JCP.0b013e3180387820 PubMed
This PDF is free for all visitors!
![]()
![]()
![]()